Notice!
I’ve found that this book project has been showing
up on more and more search engines lately and is also being directly linked to
for the information it contains(1). I therefore find it necessary to warn all
persons viewing this document that it is a work in progress, and as such it
contains errors of all kinds, be them in experimental procedures that may cause
harm, or in faulty reasoning that would get you slapped by nearly any chemistry
instructor. Please for now take
the information here with a grain of salt.
|
Most Importantly! By reading further you
agree not to hold the authors of this document responsible for any
injuries/fatalities that may occur from attempting to make any of the
products or following any of the procedures that are outlined within. Chemistry inherently possesses a degree of
danger and you must understand this, wear gloves and more if the situation
calls for it, your safety is in your own hands, not mine! |
Also note that this project is open for contribution
by any party on the internet. Simply
submit a section to Rob.Vincent@gmail.com
and it will be added into the text pending editing and such within a few weeks. Any person contributing will have their name
mentioned in the credits. Thank you for
reading this, and enjoy!
|
1 |
Although
this document may be directly linked to, it will not work in that manner as I
have hotlink protection for PDF documents, however directly linking to the
html document is possible, still though I would prefer links be to the main
book project page. |
4.0 Lab Reagent Types (Intro, discuss overlap, and generalization)
Rather then attempt to give you a chemical, name it, give you its properties, have you memorize those and move on to the next one I have organized this area to help you learn chemical properties more readily. There is a bit of generalization here and some overlap, however rather then learning the chemical then the properties, the purpose of this is to tell you the properties then give you a list of the chemicals that posses these properties along with a bit of relevant data for each. In doing this it is easier to go into deeper detail on exactly the designated title means not only under STP (Standard Temperature and Pressure) but also under extraneous conditions that you might be required to work with them at.
4.1 Acid / Base Theory (Aqueous Solution)
pH
“pH”; everybody knows the term but what does it
really stand for? And perhaps more importantly what is it good for?
pH stands
for “potential of Hydrogen” (from the original German term “potenz”). It is a measure for the activity
of hydrogen and because the activity of hydrogen in water equals the acidity of
that water the pH effectively denotes the acidity of a solution. When hydrogen
cations (H+ ions) are introduced into water they react with water to
form the hydronium ion (also referred
to as the oxonium ion) which is
denoted as H3O+(aq). The hydronium ion is the ion that
gives acidic solutions their acidic nature. The direct opposite of the
hydronium ion is the hydroxide ion (denoted as OH-) which makes
water alkaline. Water always contains H3O+ ions and OH-
ions (hydroxide ions) but in pure water they are in equilibrium which means
they cancel each other out for as the acidity of the water is concerned. When
an acid is added to the water the equilibrium shifts to the acidic end of the
spectrum which means more H3O+ ions are present in the
solution than OH- ions. When a base is added the equilibrium shifts
to the alkaline end of the spectrum which means more OH- ions than H3O+
ions are present in the solution. A pH from 0 to 7 means the solution is
acidic (so more hydronium ions than hydroxide ions); a pH of 7 means the water
is neutral (there are as many hydroxide ions as there are hydronium ions
present in the solution) and a pH from 7 to 14 means the solution is alkaline
(more hydroxide ions than hydronium ions).
The highest
attainable pH at STP (standard temperature and pressure) is 14 and the lowest
attainable pH at STP is 0. When the temperature and pressure do not conform
perfectly to STP the minimum and maximum pH will vary accordingly. This is
however not essential knowledge for hobby-chemists and as such I will not go
into it any further.
Two kinds of acids and two kinds of bases.
There are
two kinds of acids and bases; strong and
weak. The strong versions do not
form equilibriums in water but simply completely dissociate. The weaker
versions will form an equilibrium in water and as such they will generally not
be nearly as acidic or alkaline as the strong version.
|
Strong acids |
In solution |
|
Hydrochloric
acid (HCl) |
H3O+(aq)
+ Cl-(aq) |
|
Sulphuric
acid (H2SO4) |
H3O+(aq)
+ HSO4-(aq) |
|
Nitric
acid (HNO3) |
H3O+(aq)
+ NO3-(aq) |
|
Perchloric
acid (HClO4) |
H3O+(aq)
+ ClO4-(aq) |
|
Hydroiodic
acid (HI) |
H3O+(aq)
+ I-(aq) |
|
Hydrobromic
acid (HBr) |
H3O+(aq)
+ Br-(aq) |
|
Weak acids |
In solution |
|
Ethanoic
acid (Acetic acid) |
C2H3OOH(aq) |
|
2-hydroxy-1,2,3-propanetricarboxylic
acid (Citric acid) |
C3H4OH(COOH)3(aq) |
|
H3BO3
(Boric acid, aka B(OH)3
) |
H3BO3(aq) |
|
HF (Hydrofluoric acid) |
HF(aq) |
|
H3PO4
(Phosphoric acid) |
H3PO4(aq) |
The first
difference that catches the eye is the fact that strong acids are denoted as
free constituent ions and that weak acids are denoted as {weak acid}(aq). This is
because the strong acids always
completely dissociate in water whereas the weak acids only very partially
dissociate in water.
|
Strong base |
In solution |
|
KOH
(Potassium hydroxide) |
K+(aq)
+ OH-(aq) |
|
NaOH
(sodium hydroxide) |
Na+(aq)
+ OH-(aq) |
|
Ca(OH)2
(calcium hydroxide) |
Ca2+(aq)
+ OH-(aq) |
|
Ba(OH)2
(barium hydroxide) |
Ba2+(aq)
+ OH-(aq) |
|
Na(C2H5O)
(sodium ethoxide) |
Na+(aq)
+ OH-(aq) + C2H5OH(aq) |
|
Weak base |
In solution |
|
K2SO4
(potassium sulfate) |
2K+(aq)
+ OH-(aq) + HSO4-(aq) çè 2K+(aq) + SO42-(aq) |
|
K2CO3
(potassium carbonate) |
2K+(aq)
+ OH-(aq) + HCO3-(aq) çè 2K+(aq) + CO32-(aq) |
|
KF
(potassium fuloride) |
K+(aq)
+ OH-(aq) + HF(aq) çè K+(aq) + F‑(aq) |
|
Na2HPO4
(sodium biphosphate) |
Na+(aq)
+ OH-(aq) + H2PO4-(aq) çè Na+(aq) + HPO42-(aq) |
|
NH3
(ammonia) |
NH4+(aq)
+ OH-(aq) çè NH3(aq) |
The obvious
difference here is that in the case of the weak bases they are denoted as
equilibriums when in solution whereas the strong bases (which do not form equilibriums)
are denoted simply as their respective constituent ions in solution (so with
“(aq)” at the end).
Calculations.
The pH of a
solution can be calculated as follows:
pH
= -log[H3O+]
please note
here that [H3O+] denotes
the hydronium concentration and that the addition of the p (for potential)
effectively means you take the negative logarithm of the value without the p
(in this case negative logarithm of the hydronium concentration). So when I
take 2 moles of the “strong acid” HCl(g) and add enough water to make a
solution of 1L of water I will have a 2M solution of hydrochloric acid. The pH
of that solution would be –log(2) = -0.30 M = -3 * 10-1. Which not
only means the solution would be rather acidic but also immediately shows that
the pH of a solution might be realistically expected to be between ~ -1,5 and ~
15,5 rather than between the limits that are predicted by the rule. Don’t worry
it really doesn’t make much of a difference when you’re using these formulae.
An
important conclusion that can be drawn from this formula is that one can easily
calculate the hydronium concentration (and as such the amount of hydrogen
cations in solution) by means of the following calculation:
10-pH
So when a
solution has a pH of 3,7 the [H3O+] = 10-3.7 =
0,0001995 = 2 * 10-4 mole/L
(can also be denoted as 2 * 10-4
M).
Ka
and pKa
The degree
to which an acid will dissociate in water is denoted by means of its Ka. The Ka is basically the dissociation
constant of the acid in water. Ka’s can be denoted in a simpler form by taking
their negative logarithm to yield the pKa.
![]()
pKa = -log(Ka)
These
formulae can be used to calculate the amount of hydrogen cations and the and
the pH of a solution of a certain strong acid. The calculations for a base can
be performed in the same way but using the Kb in stead and the result of the
calculation would yield the [OH-] which can be transformed into the
pH by means of the following formulae:
-log[OH-] = pOH
14,00 – pOH = pH
This is
because pH + pOH for a certain solution at STP must always equal 14,00. Also
the Ka of a base can be calculated by means of the following formula:
pKa + pKb = pKw (which means KaKb = Kw = 10-14)
pKw = 14 (which means
Kw = 10-14)
10-pKa =
Ka
To
calculate the pH of a solution of a weak acid (or the pH of the solution of a
weak base) the following formulae can be applied:
![]()
This is
because only a small portion of the weak acid will dissociate. You can calculate
by calling [X-] and [H3O+] x and filling out
the equation. Some simple calculus should yield the [H3O+].
It is useful to have a calculator handy in that case because it will be rather
laborious to calculate x.
E.g. a
0,1M solution of formic acid (HCOOH)
pKa = 3,79 è Ka = 1,6 * 10-4
[HCOOH] =
0.1
Fill out
the formula, do the math and the answer will prove to be 3.9 * 10−3.
(hence the pH will be ~2,4)
The most loose definition of acids that most people are familiar with defines an acid as a chemical that is able to donate a hydrogen cation to water. In doing this it generates the H3O+ cation which is the acidic component of water. However this does not cover every acid under every circumstance by a long shot. Never the less, water is a common solvent and in defining an acid it is easier to use definitions governed by water then add in the exemptions later for non aqueous systems.
|
Common Acids |
|
Acetic Acid H3COOH |
Commonly known as vinegar, this acid forms no confirmed azeotrope with water. It is somewhat strong in concentrated form, dissociating to an appreciable extent. Acetate salts are usually soluble and are therefore a good source of metal ions in solutions, however solutions are slightly basic. |
|
Hydrochloric Acid HCl (Muratic Acid) |
Sold as a solution in water of HCl gas, hydrochloric acid is a strong mineral acid. The commonly available forms are 20% (The azeotrope), 38% (concentrated with a density of 1.19 g/cm3) It will attack anything in the reactivity series above hydrogen, most chlorides are at least slightly soluble. |
|
Nitric Acid HNO3 |
Not commonly available, which is a shame considering how useful it is. Nitrates of metals are all soluble so it provides a good ability to solvate a cation of your choice. It is a strong oxidizing acid, able to oxidize metals readily at room temperature evolving nitrogen oxides. |
|
Sulfuric Acid H2SO4 |
The staple acid of at home chemistry. Very difficult to obtain in some countries but relatively easy to find in America. Most sulfates are soluble in water although there are some notable exceptions (CaSO4, BaSO4, and PbSO4). Concentrations vary widely form common battery acid (~30%) to additional acid anhydride dissolve in 100% H2SO4. Weakly oxidizing. |
|
Sulfamic Acid NH2SO3H |
Somewhat readily available and stronger then many organic acids. It forms may highly soluble salts, in the pure form it is a solid. |
|
Boric Acid B(OH)3 |
Very weak acid. Borates are readily available in the cleaning industry, dehydrates easily to boric oxide. Boric acid readily forms boric esters, which burn to give beautiful colors. |
|
Hydrofluoric Acid HF |
Weak solutions are available over the counter for cleaning rims of cars and such (~3%). Hydrofluoric acid is very toxic and highly concentrated solutions can kill very rapidly if splashed on the skin. As an acid though it is somewhat weak compared to hydrochloric, as a pure compound it is a liquid near room temperature. |
|
Cyanuric Acid HOCHC(OH)NC(OH)N*2H2O |
Somewhat weak acid available for adjusting the pH of pools. |
|
Phosphoric Acid H3PO4 |
Concentrated phosphoric acid is a fairly strong acid. It readily attacks metals forming phosphates, which are on the whole soluble in water. The more concentrated the solution the more syrupy it is until it becomes a solid. Phosphoric acid will not boil, it will continuously loose water even past where it is 100% dehydrating to other forms of phosphoric acid such as pyrophosphoric acid. Avalible for cleaning metal and for marine cleaning. |
Time to introduce you to the metal activity series. Although important to other chemistry concepts it answers one question regarding acids that people ask most often. “What will an acid dissolve?” Below is a list of elements, towards the end of the list is hydrogen, and anything to the left of it will dissolve to some extent in acid. Those in the lighter color to the immediate left dissolve slowly-very slowly, going to the darker color even more to the left we find elements that will not only displace hydrogen from an acid but will react with steam. Finally those furthest to the left will readily react with water and their subsequent reaction with acid would only be described as intensely violent. This is a standard activity series, some series will have elements in slightly different relation to one another but this is the basic order.
Li K Ba Ca
Na Mg Al Mn Zn Cr Fe Cd Co Ni Sn Pb (H2)
Cu Ag Hg Pt Au
 So you’re
looking at the list and you wonder, “What about those elements to the right of
hydrogen?” A good question, those
elements will not displace hydrogen from acid and as a consequence they could
be considered inert in that respect.
But that would be a mistake to assume they would remain inert in all
respects. There is a way around this
inertness, the addition of an oxidizing agent.
The principle, let’s say for example you have a piece of copper that has
some surface oxidation, now let’s say you put it into some hydrochloric acid,
immediately the oxidized layer dissolves off tinting the acid a green/blue
color. Pulling out the copper it looks
fresh and clean, no oxidation. So, the
oxidized layer dissolved, if you were to leave it out the oxygen in the air
would re-oxidize that top layer, you could dip it back into the hydrochloric
acid, and dissolve yet more of the copper.
In this case the atmospheric oxygen is the oxidizing agent, bubbling air
though HCl while dissolving copper accomplishes this. But another way would be to add an oxidizing agent to your acid,
an even better way would be to have an oxidizing acid. Perchloric acid (HClO4) and
nitric acid (HNO3) are both oxidizing agents as well as acids [Hot
concentrated H2SO4 is also an oxidizing agent], as a
matter of fact nitric acid almost always functions as an oxidizing agent unless
coupled with a very reactive metal, magnesium will actually liberate hydrogen
for the first few seconds of reacting with nitric acid but after that it will
be preferably oxidized first. Oxidizing
acids will not dissolve some elements that have insoluble oxides, the formation
of the oxide forms a protective layer pacifying the metal to further attack, a
good example is aluminum in concentrated HNO3, also tin can be pacified in this
way under some conditions.
So you’re
looking at the list and you wonder, “What about those elements to the right of
hydrogen?” A good question, those
elements will not displace hydrogen from acid and as a consequence they could
be considered inert in that respect.
But that would be a mistake to assume they would remain inert in all
respects. There is a way around this
inertness, the addition of an oxidizing agent.
The principle, let’s say for example you have a piece of copper that has
some surface oxidation, now let’s say you put it into some hydrochloric acid,
immediately the oxidized layer dissolves off tinting the acid a green/blue
color. Pulling out the copper it looks
fresh and clean, no oxidation. So, the
oxidized layer dissolved, if you were to leave it out the oxygen in the air
would re-oxidize that top layer, you could dip it back into the hydrochloric
acid, and dissolve yet more of the copper.
In this case the atmospheric oxygen is the oxidizing agent, bubbling air
though HCl while dissolving copper accomplishes this. But another way would be to add an oxidizing agent to your acid,
an even better way would be to have an oxidizing acid. Perchloric acid (HClO4) and
nitric acid (HNO3) are both oxidizing agents as well as acids [Hot
concentrated H2SO4 is also an oxidizing agent], as a
matter of fact nitric acid almost always functions as an oxidizing agent unless
coupled with a very reactive metal, magnesium will actually liberate hydrogen
for the first few seconds of reacting with nitric acid but after that it will
be preferably oxidized first. Oxidizing
acids will not dissolve some elements that have insoluble oxides, the formation
of the oxide forms a protective layer pacifying the metal to further attack, a
good example is aluminum in concentrated HNO3, also tin can be pacified in this
way under some conditions.
|
Oxidizing Acids By being an oxidizing agent the acid must simultaneously be reduced in the reaction. Therefore when copper is subjected to the action of nitric acid copper is oxidized and the nitrate anion is reduced to any of a number of nitric oxides depending on the conditions under which the oxidation took place. Here are some examples of the reactions of nitric acid: 2HNO3(aq) + Mg(s) Þ Mg(NO3)2(aq) + H2(g) Rarely occurs, only happens initially with magnesium or even more reactive metals [Na, K, Li, etc.], not important. 3Cu(s) + 8HNO3(aq) Þ 3Cu(NO3)2(aq) + 4H2O(l) + 2NO(g) This is an example of nitric acid acting as an oxidizing agent when dilute. Cu(s) + 4HNO3(aq) Þ Cu(NO3)2(aq) + 2H2O(l) + 2NO2(g) This is an example of nitric acid acting as an oxidizing agent when concentrated. Notice the ratio of nitric acid molecules reacting with copper compared to the dilute reaction above. [Note, a picture of this reaction is shown in the picture in the preceding section] P4(s) + 20HNO3(aq) Þ 4H3PO4(aq) + 20NO(g) + 4 H2O(l) Concentrated nitric acid can also oxidize elements such as phosphorus, silicon, sulfur, and occasionally carbon, especially when heated. Fe(s) + 6HNO3(aq) Þ Fe(NO3)3(aq) + 3 H2O(l) + 3NO2(g) When metals capable of multiple oxidation states are dissolved in concentrated nitric acid they will usually take the highest normal oxidation state, in this case iron becomes +3 in preference to +2. Similarly, when copper or mercury, some of the more reactive of the metals that follow hydrogen in the activity series, come into contact with hot concentrated sulfuric acid they can be oxidized and the sulfuric acid reduced. Cu(s) + 2H2SO4(l) Þ 2H2O(l) + SO2(g) + CuSO4(aq) Perchloric acid is encountered to a considerably lessened extent in the laboratory, it has a nasty reputation for exploding for no reason, generating out of control reactions, creating fire hazards, and making unstable salts. |
Another thing to consider when pondering weather a metal will dissolve
in an acid is weather the salt formed would be soluble. One would not logically think that silver
would dissolve in hydrochloric acid independent of its unreactivity simply
based on the fact that the silver chloride thus formed is totally insoluble. Even a piece of barium metal tossed in H2SO4
may become pacified which is an amazing thing considering it would react very
rapidly with water. Oxidizing ability
aside there is another method to measure the strength of an acid, the pH scale
and the pKa scale, which were discussed in the opening section.
4.3 Bases

As shown in the above picture bases can rapidly attack some metals just as acids can. To the left in the above picture some aluminum turnings have been placed into a weak potassium hydroxide solution, to the right a weak acid solution is also attacking a similar amount of aluminum. Hydroxides can attack a number of metals, especially when hot and concentrated, however the reactivity shown with aluminum, zinc, and magnesium can be considered special cases for the common metals.
|
Common Bases |
|
Sodium Hydroxide NaOH |
Avalible over the counter as lye, sodium hydroxide serves the purpose of being the no-nonsense base, addition of sodium hydroxide to an aqueous solution automatically increases the hydroxide ion concentration and brings only the sodium cation along with it. |
|
Sodium Carbonate Na2CO3 |
Sodium carbonate is available as “Washing soda” it is usually the decahydrate (*10H2O) but that does not interfere with calculations as long as it is accounted for. Be wary of other impurities though. Sodium carbonate is a great base because the reaction with acidic components is driven foreword strongly by the loss of carbon dioxide from solution. |
|
Sodium Bicarbonate NaHCO3 |
Less basic in solution then sodium carbonate but still able to neutralize acids well. It is safer on the skin and is therefore the choice base to have laying around in case of an acid spill. |
|
Ammonia NH4OH |
Ammonia gas can simply be bubbled into solution to increase its pH. That is a great advantage to ammonia. Also it can be forced from solution after its purpose has been served, the gas itself will react with acids even if they are not aqueous either. |
|
Trisodium Phosphate Na3PO4 |
Basic in water solution due to the equilibrium present between the phosphate anion and the hydrogen phosphate anion and the dihydrogen phosphate anion which take up hydrogen from the water and therefore leave hydroxide anions. This base is available as prills for a stripping agent in painting. |
 The case to the left shows the effect of hydrobromic acid on hydrogen
peroxide. Whereas acidic peroxide solutions are one of the possible
oxidizing agents that one can pick from, using hydrobromic acid/H2O2 solutions
is not advisable. The hydrobromic acid will act as a catalyst to decompose
the H2O2 resulting in lessened yields, and in addition, the oxidation potential
of the mix is enough to oxidize Br- anions to elemental bromine. This is
clearly shown, initially the H2O2 and the HBr solutions were clear, when mixed
they immediately turned yellow, and upon standing for a minute or so the mix
was a deep red with bromine vapors clearly stagnant above it. Just goes
to show you that you need to consider even the smaller things when attempting
oxidation reactions.
The case to the left shows the effect of hydrobromic acid on hydrogen
peroxide. Whereas acidic peroxide solutions are one of the possible
oxidizing agents that one can pick from, using hydrobromic acid/H2O2 solutions
is not advisable. The hydrobromic acid will act as a catalyst to decompose
the H2O2 resulting in lessened yields, and in addition, the oxidation potential
of the mix is enough to oxidize Br- anions to elemental bromine. This is
clearly shown, initially the H2O2 and the HBr solutions were clear, when mixed
they immediately turned yellow, and upon standing for a minute or so the mix
was a deep red with bromine vapors clearly stagnant above it. Just goes
to show you that you need to consider even the smaller things when attempting
oxidation reactions.
|
Common oxidizing agents |
|
Potassium Perchlorate KClO4 |
Solid, white powder, non-hygroscopic, very slightly soluble in water, usually has to be bought from a pyrotechnics supplier or made via electrolysis. |
|
Sodium Nitrate NaNO3 |
White powder soluble in water, hygroscopic, slightly saline/bitter taste (don't taste it!). Acid solutions will attack noble metals such as copper. Occasionally available during the summer months as fertilizer. |
|
Nitric Acid HNO3 |
Clear - Yellow/Green liquid. Available in various concentrations, >70% show remarkable oxidizing capabilities, lower concentrations available over the counter for hydroponics. |
|
Hydrogen Peroxide H2O2 |
Clear liquid, available in various concentrations from 2% to 99% solutions greater then 50% should be treated with care as combination with many things can cause them to explode. Greatly attacks tissue. |
|
Potassium Dichromate K2Cr2O7 |
Bright orange solid, soluble in water. Solutions of potassium dichromate with sulfuric acid were once one of the most routine things to clean lab glass with. Potassium dichromate is considered carcinogenic. |
|
Sodium Hypochlorite NaClO |
Clear-Yellow/Green liquid strong chlorine type smell. Surprisingly good widely available oxidizing agent. Considerably more powerful in concentrations greater then 12.5% and especially when hot. |
|
Sodium Chlorate NaClO3 |
White solid available as a weed killer in some areas. Toxic and hygroscopic it has powerful oxidizing powers as a solid, when heated on its own it undergoes self oxidation-reduction to perchlorate and chloride. |
|
Potassium Permanganate KMnO4 |
Bright purple solid possessing great oxidizing ability as a solid and in either basic or acidic solution. Found as a treatment for water in areas where iron is a problem. |
Aqueous Oxidations:
Hydrogen Peroxide Solutions

Shown above is an attempt to dissolve nickel metal under various conditions. Although not totally apparent the HCl solution and the H2SO4 solution showed little attack. The HCl/H2O2 solution did show some attack. However it was the H2SO4/H2O2 solution that showed incredible results. As you can see the entire top of the nickel in the test tube to the far right has eroded to a point. In addition the whole bottom of the test tube is full of nickel (II) sulfate crystals. The mix of H2O2 with H2SO4 also looks entirely different from just H2SO4 acting alone, seen in the second to left picture, a cloudy mixture formed in that instance unlike the superb green mixture formed from H2SO4 reacting in tandem with H2O2. The reason for this is H2O2 increases the process by oxidizing the noble metal, once the surface is oxidized the oxide dissolves in the acid, and once it dissolves in the acid the H2O2 can oxidize the surface again.
4.4a Molten Salt Oxidations /
Solid State Oxidations:

With molten salt oxidations one can force metals into oxidation states that would be very difficult to achieve in the aqueous phase and would be considerably less stable if formed in that way as well. The actual chemistry of such oxidations is usually complex but there are only two simple needs to perform most of these oxidations, an alkali metal hydroxide, and an oxidizing agent usually an alkali metal nitrate where the gaseous visages of the oxidizing anion might readily leave the melt and the remaining cation will not interfere. Several reactions go on in such melts, but mixtures involving potassium hydroxide make a good example:
2KOH Û H2O + K2O
4KOH + 3O2 Û 4KO2 + 2H2O
Now, peroxides and superoxides are strong oxidizing agents in their own right, but the oxide anion O2- is incredibly basic as is the superoxide O-1/2 (note that potassium peroxide is fairly unstable, and does not exist appreciably in the molten state). The oxidizing agent in the melt, usually something like potassium nitrate helps to drive this equilibrium, acting as a very convenient source of oxygen. At these temperatures things like potassium nitrate are very reactive, a cotton glove for instance, coming into contact with molten KNO3 will burst into flames, but at room temperature KNO3 could be safely handled with ones bare hands. All in all, the very basic electron rich environment can best stabilize a number of high oxidation state compounds, which can then be used in further chemical endeavors.
The melts used for these oxidations are fairly corrosive; vessels of nickel, platinum, and silver are best. Glass is out of the question, as the strong bases present will attack it. However for the roughest oxidations, disposable vessels of steel or commonly available pipefittings can work. These will contaminate products obtained but compounds formed under these conditions are not going to be very pure anyways. Here are a few examples of high oxidation state compounds that can be made by these methods:
Ferrate [FeO4]2- : Ferrates will decompose almost instantly if in acid solution, quickly in neutral solutions, and slower in basic solutions. Kept free of moisture and stored without access to air, ferrates will keep for several weeks or months. They are made by fusing ferric oxide with KOH and an oxidizing agent and are purple/red in color. Ferrates can be precipitated from an aqueous solution as the slightly soluble barium salt or by concentrated potassium hydroxide solution.
Bismuthate [BiO3]- : Bismuthate as with many other high power oxidizing agents is available from chemical suppliers usually only as a purity of 85% or so, further refinement being unnecessary due to some of the brute force type oxidations done with it. Out of these four this is the second most commercially available oxidizer listed. It can oxidize manganese ions in solution to permanganate and is usually found for sale as the sodium salt. It can be prepared by fusing bismuth trioxide with potassium hydroxide as long as the mix is exposed to air.
Chromate [CrO4]2- : This is the most widely available oxidizer listed here. Chromates are toxic and should be handled with care. They are also the weakest oxidizer on this list. They are usually formed by fusing chromium (III) oxide, acidification of a solution of chromate will lead to the formation of dichromate which will usually precipitate if the concentration is high enough and the temperature lowered afterward, dichromates being more useful then chromates. Industrially this process is used to make dichromate by fusing chromite ore (FeCr2O4) with potassium hydroxide in the presence of oxygen, after oxidation the mixture is dissolved in water and acidified, the chromate being converted to dichromate and the ferrate going to soluble Fe3+.
Manganate [MnO4]2- : Manganates are green in color and the product of fusing manganese dioxide. They are fairly unstable and upon addition to water and acidification yield a solution of permanganate. Subsequent filtering and crystallization allowing for the production of permanganate at home, which is useful as well.
When an oxidation is completed there are two courses of action, if the product is stable to the atmosphere it can be poured onto a sheet of steel and allowed to cool quickly, then broken with a hammer and stored. However if it is not then it must be covered in the crucible while covered and once cooled chipped out and stored. In either case these melts are very strong oxidizing agents and must never come into contact with anything organic or flammable. In addition these melts can NEVER be poured directly into water while in the molten state as they can very often explode. Temperature control is not a major issue with most molten oxidations but things should still never be heated too strongly, molten oxidations work best in the range from 375 – 550 °C and the amount of time to hold the reactants there depends strongly on what you are trying to oxidize and the amount you have in the mixture. If you desire to produce an oxidizing agent that is very unstable at high temperatures considering an eutectic mixture can help greatly, a 50/50 mixture of NaOH/KOH has a significantly lower melting point then either component alone.
Aside from the inherent risks of holding oxidizing mixtures at high temperatures, the use of nitrates can lead to the formation of nitrogen oxides which are extreme hazards therefore the actual heating step should be preformed while you are not in the company of the reaction vessel. Additionally the subsequent dissolution and acidification of some of these mixtures can also lead to nitrogen oxide release if the reaction yielded a large amount of nitrites as can often be the case. Not to mention that the oxidation reactions also have the possibility of going awry, if excessive frothing or sparks start to come from a reaction mixture that is your key to exit the area. One final note, chlorates and perchlorates can be used for these oxidation reactions, however acidification of a solution of chlorate can yield explosive quantities of chlorine dioxide and additionally the reaction itself can run away in the presence of certain metal oxides.
The most common class of reducing agents one runs across are usually the active metals. In pyrotechnics aluminum, magnesium, and occasionally zinc are used with strong oxidizing agents such as perchlorates and nitrates to give spectacular exothermic reactions. However on a more controlled level these metals can also be used to give reliable reactions even in reactions involving aqueous reactants. Additionally there are a number of organic reducing agents which are most popular in the field of organic chemicstry. Both organic and strictly inorganic reducing agents are of great utility in the chemistry lab however they are more difficult to obtain then oxidizing agents in most cases and in the case of the metals, usually difficult to get into a workable form.
|
Reducing Agent |
Source |
Example of Use |
|
Aluminum Al |
Pyro suppliers, scrap yards, foil |
Powdered aluminum is a powerful solid state reducing agent (Thermite reactions), it can also work to reduce cations in the aqueous phase and can be amalgamated with mercury for organic reductions. |
|
Magnesium Mg |
Scrap yards, camping suppliers (fire starter), pyro suppliers, cell phone pieces and in some other high end applications (bike frames etc.) |
Powdered magnesium is a stronger reducing agent then aluminum, it reacts slowly with water reducing it. Magnesium can reduce a number of inorganic compounds such as NaOH works in thermite type reactions. |
|
Hydrogen H2 |
Reaction of a strong mineral acid with an active metal (aluminum, magnesium, iron) |
Reductions with hydrogen gas usually take place at elevated temperatures when concerning inorganics and under high pressures with organics, it is very useful but difficult for an amateur to use. |
|
Lithium Li |
Purchased from a chemical supplier, some batteries contain lithium, home electrolysis in non-aqueous medium or of electrolysis of lithium chloride/bromide eutectic. |
Lithium metal finds use either alone or in compounds for the reductions of organic compounds, lithium dissolved in ammonia or n-butyl lithium being incredibly strong organic reducers. |
|
Sodium Na Potassium K |
Purchased from chemical supplier, made at home via electrolysis or reduction of salts. |
Considering the high reactivity for each of these they find little practical use in but are occasionally called upon. A liquid eutectic is formed between these two elements with is unbelievably reactive. |
|
Carbon C |
Graphite, sugar carbon (formed by heating sugar till it decomposes), coal, charcoal |
Carbon is an excellent reductant however its use requires very high (>900C) temperatures. |
|
Sulfite SO32- |
Some OTC sources, bubbling sulfur dioxide into basified water. |
Sulfites are oxidized to sulfates and in the process function as weak reducing agents. |
|
Citric (Ascorbic) acid C6H8O7*H2O |
Vitamin C tablets, sold as citric acid for flavoring, extracted from lemon juice |
Weak aqueous reducing agent, works better at higher temperatures, good for making metal powders of somewhat nobel metals (e.g., nickel, chromium, silver, etc.) |
|
|
|
|
Runaways can happen with reductions as well as oxidations.Chemistry can be fun and educating in many ways but one should always remember to be careful. Sometimes one just forgets to do the background work first, before moving to do the actual experiment.. That happened to me some time ago. I had previously prepared few moles of nitrotoluene and planned to reduce some of it to toluidines. So, I remembered the standard Sn and Fe reductions of aromatic nitro compounds with HCl and did a few calculations on the amounts of reactants required. Ended up using 1mol of nitrotoluene and the appropriate amounts of 40 micron, hydrogen reduced Fe powder and 37% aqueous HCl. The nitrotoluene was mixed well with the Fe powder and a little distilled water in a 500ml erlenmeyer flask. I was first going to use a magnetic stirrer to efficiently keep the iron powder suspended but remembered that iron is magnetic, luckily before I dumped in the stirbar.. HCl was put into an addition funnel and was added drop by drop to the mixture, which I swirled continuously. I expected a fast temperature rise but there was none. HCl addition was continued a bit faster and then the temp finally started to rise. Stopped the addition for a moment and swirled strongly. After continuing the addition the temp didn't rise much more so I decided that I’d add half of the remaining HCl now and the rest after ten minutes or so. That was big mistake. Soon the temp started to rise fast. I swirled the flask as strongly as I could but it didn't seem to help. I took the thermometer off as it was nearing the limit (100C). I put the flask in to an ice bath and swirled vigorously. The flask started to feel _very_ hot at that time and I was ready to dump it in the bath, but I was too late. Suddenly the mix started to boil and shoot itself out of the flask. I had to let go off the flask as it was so hot and then the reaction got so vigorous that it shot all the remaining liquid out in a geyser like fashion on the floor. I quickly took a bottle of water and poured it in and over the flask. After I had the reaction tamed I decided to go outside and take the annoying gasmask off for a while. I immediately smelled the slightly irritating smell of nitrotoluene and realised that all the reaction mixture had probably boiled out of the flask with the steam.. The whole neighbourhood smelled of nitrotoluene for a few hours, as it wasn't windy. The flask seemed ruined but I got it eventually cleaned with some HCl and sodium ethoxide solution. The worst part was cleaning the mixture off the coarse concrete floor.. And the smell stayed for ages in my lab.
That certainly thought me a lesson to always find about different reactions and the possible mistakes that could be made during it. And not to start doing a new reaction with that much material. Starting from mill moles is much more recommended to get a feel for the reaction, before scaling up! |

4.6 Dehydrating Agents/ Desiccants

In the case of the above picture nickel chloride is shown. The kernel on the right being an anhydrous lump, and the green solution on the left being the same amount solvated in water. This is just one example of a hygroscopic salt that changes color when hydrated. Mind you, by being hygroscopic, a salt is not at the same time disquecent. A disquencent salt will pull enough water from the air to put itself into a solution, an example being NaOH or CaCl2, a salt that is hygroscopic, but not disquecent, will form a stable solid hydrate that is more easily handled. Another example of a color changing salt that forms a stable hydrate is copper sulfate, which is colorless when anhydrous but turns blue when its water removing capacity has been used up, it can be reactivated for use by heating for an extended period of time.
The main use of desiccants is to remove water, usually from a liquid or gas to render that liquid or gas largely free of water in reactions where water might inhibit a desired reaction, interfere with a reaction, or cause extraneous byproducts. Drying agents can form very strong bonds to water, strong enough to take water from a chemical bond, such as the reaction between concentrated sulfuric acid and sugar, where the sulfuric acid will remove the water from the molecule C6H12O6 leaving behind just carbon in a very pleasing visual display, phosphorus pentoxide, and hot NaOH are the only other drying agents on this list that posses this strength of drying power, agents of this type are referred to as dehydrating agents.
|
Chemical Name /Formula /Formula of hydrate |
Form of anhydride / Form of hydrate |
Details on Agent |
|
Sulfuric Acid H2SO4 H2SO4*xH2O |
Dehydrating Acid Heavy Liquid |
Heavy liquid, dehydrating action most apparent at high concentrations 90%+, concentrations higher are possible by dissolving the acid anhydride (SO3) in concentrated H2SO4, such solutions (called oleum) possess additional dehydrating strength, but remains liquid, will dehydrate sugar to carbon. Great for drying liquids. |
|
Phosphorus Pentoxide P2O5 H3PO4 or H3PO3 |
Solid/Powder (becoming plastic like/liquid) Dehydrating Agent |
Solid/Powder formed by burning phosphorus in air. Disquecent, pulling water from air making a crust on the surface forming differing phosphor acids, phosphinic acid, phosphoric acid, etc. Very strong dehydrating agent, forms N2O5 from concentrated HNO3. |
|
Magnesium Sulfate MgSO4 MgSO4*7H2O |
Drying Agent Solid Forms stable hydrate |
White powder, commercially the heptahydate *7H2O is available, however this can be dehydrated in an oven maxed out for a few hours. There is no color change upon hydration or dehydration. MgSO4 is cheap and decent for drying some gasses and liquids however its action is not very strong. The hydrated salt is a solid. |
|
Calcium Chloride CaCl2 CaCl2*2H2O (But will go further) |
Drying Agent Solid (anhydrous) Liquid (hydrated) |
CaCl2 is widely available for use as deicing or as a drying agent for use in basements. It comes in the form of solid prills that will suck moisture from the air until they turn into a puddle. The drying action of this solid is similar to anhydrous MgSO4 but the liquid hydrated state may run back into reactions. |
|
Copper Sulfate CuSO4 CuSO4*5H2O |
Drying Agent Solid (Colorless) Green/Blue when hydrated |
Widely available for killing roots in sewer lines or preparable by dissolving copper in hot sulfuric acid, it can be made anhydrous by heating. Solid that changes color when its drying action is used up. Good for dying alcohols and such, action is stronger then CaCl2 or MgSO4 and it is regenerateable over high heat. |
|
Calcium Sulfate CaSO4 CaSO4*2H2O |
Drying Agent Solid White Forms a stable hydrate |
Available over the counter as the semi-hydrated ‘Plaster of Paris’ or as drywall, which is the dihydrate. Made anhydrous by heating is possesses decent drying abilities but its ready availability and low cost make it somewhat desirable, color changing versions are available from chemical supply companies. |
|
Magnesium Perchlorate Mg(ClO4)2 Mg(ClO4)2*6H2O |
Drying Agent Solid but liquid when hydrated |
One of the kings of the drying agent world, magnesium perchlorate possesses exceptional drying ability. However it can explode when exposed to solvent vapors or intense heat and therefore it has fallen into disuse, concentrated sulfuric acid or phosphorus pentoxide often substituted for it. |
|
Sodium Hydroxide NaOH NaOH*xH2O |
Dehydrating Agent Solid but liquid when hydrated |
Available over the counter, store bought NaOH contains some impurities and is of a variable composition of sodium oxide and water. It is a good drying agent for taking the last bit of water out of liquids in which it is insoluble but does not have the ability to dry large amounts well, just small amounts of a liquid/solid/gas thoroughly. |
|
Calcium Oxide CaO Ca(OH)2 |
Drying Agent Solid forming a stable hydrate |
Widely available for adjusting the pH of soil, the calcium hydroxide thus formed is only slightly soluble, although regenerateable through heating it is more often then not simply used once. |
The activity of a dehydrating agent, the ability of it to pull water from its surroundings is not usually something to be directly gauged, but there is a great difference in each drying agents ability to pull water and trap it. For example, sulfuric acid when concentrated will char wood and turn sugar to coal. Whereas calcium chloride will do neither. These aspects can make a great difference in their usage and what reaction they may be unsuited for.

4.7 Poisonous Reagents
A poisonous reagent might be easily classified as a chemical that requires only minimal unintentional contact to cause adverse effects. Such a definition is better suited for this class then simply a chemical that can cause harm, after all, there is a lethal dose for table salt and alcohol, so it is better to only classify those chemicals that could easily cause harm to oneself thought an accident as poisonous. From here poisonous chemicals are further divided into two categories, not independent from one another. Those chemicals that are cumulative poisons, and those that are not.
A cumulative poison is a poison whose presence in the body is not immediately eliminated and it accumulates in the system, i.e., it would be easy for a person to take in more of this poison, however infrequently, then their body will expel. This can be referred to as the half-life of a substance. Examples of cumulative poisons are lead salts, fluoride, radioactive strontium, and others. Cumulative poisons can also have almost no half life in the body, but are instead cumulative in the effects they cause, long term lung damage can result from inhalation of even minute amounts of some chemicals and upon repeated exposures that damage might become severe enough to cause emphysema or other conditions.
One mistake people take into account when handing a potentially poisonous substance and assessing its lethality is to consider the time frame over which it is lethal. Poisons that can kill in minutes such as hydrogen sulfide and hydrogen cyanide are often viewed with considerable more trepidation then other gasses like nitrogen dioxide, simply because nitrogen dioxide may not kill instantly. It still caries with it significant danger, all three of them do, and it is not a matter of will it kill you instantly or eight hours from now, it’s a matter of if the chemical you are dealing with is dangerous and taking the necessary precautions to ensure that should an accident happen the least harm will befall you. A poison is a poison, and aside from physical differences they should all be treated with the same careful consideration.
Let’s say, for example you have dissolved silver in excess nitric acid and currently have the beaker sitting in the middle of an open table. Now, you have chosen to precipitate the silver and simultaneously neutralize the excess nitric acid with sodium carbonate. You make your carbonate into an aqueous solution and add it drop wise slowly to avoid excess spattering. The next day you wake up from a restful sleep to find that your arms are covered with tiny black dots, and so is your face. Invisible drops of silver chloride solution were thrown from the beaker, carried by the wind, and otherwise deposited on your person. Had that been a highly toxic chemical you actually may have been beginning to feel the effects, a similar neutralization of a barium salt solution with excess carbonate may well make you sick within a few hours.
Always pay careful attention to chemicals that you work with that may be poisonous. And do not use them unless you feel you have to. Vapors can travel surprisingly far, heating poisonous solids may release similar vapors, some reactions may cause the breakdown of chemicals into more poisonous alternatives. The best ways to deal with a poisonous substance depends on its current form.
My poisonous substance is a liquid or is in solution:
If your substance is in water, its poisonous ability is slightly lessened, water has a weak ability to penetrate, chemicals that do however are lipophilic. Examples are carbon tetrachloride, DMSO, ethanol, ether, poisons solvated in these pose a greater hazard then those chemicals alone usually. However, should your substance be a liquid that is comprised of heavy metals directly bonded to carbon, organo-metiallic compounds, I cannot stress enough the danger involved, these will give heavy metals the most direct path into your body and straight to your brain. Wear gloves and don’t cause the solution to foam, avoid heating of solutions that contain liquid poisons unless they are completely enclosed within a glassware setup.
My poisonous substance is a solid:
As long as the substance is totally dry, if it gets on your skin it can be gingerly wiped off and the area afterward washed with copious amounts of water. Always be careful of handling poisonous solids like cyanides in windy areas or using heavily powdered derivatives which may take flight even without the presence of wind. Keep track of weighing paper and such that comes into contact with your substance and be sure to use powder funnels for the transfer to keep it off the lips of containers.
My poisonous substance is a gas:
The most dangerous form of a poisonous substance. A gas mask is recommended should you have access to cartridges that effectively filter out the gas. Look to the section on gasses (4.10) for further information. Remember that even if your mask filters the dangerous component your neighbors will have to live without a mask and are at risk as well. Always consider your environment in these reactions. Note that not all gasses have smells to indicate their presence and they can be generated in large amounts should a liquid solution containing them is heated.
Common Solvents:
Specific Solvents: (Red = Flammable; Blue =
Non-Flammable; Green = Burns with difficulty)
Density of the solvent is listed immediately
following the solvent formula.
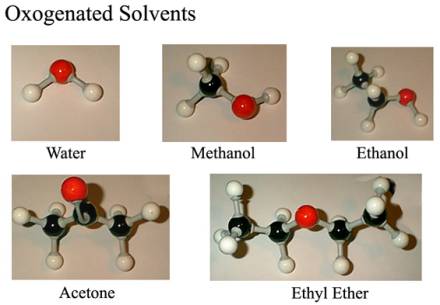
Water H2O: [1.0 g/ml] This is it, your
ace in the hole. Water is by far the
most useful, cheap, and widely available solvent you have. Known as the universal solvent, water also has
the very convenient liquid range from 0 °C to 100 °C,
it has a few somewhat painful points that become apparent when trying to remove
waters of hydration or working with hygroscopic materials but the good
definitely outweighs the bad. Water
will solvate most ionic compounds, as the saying goes, like dissolves
like. Other solvents with properties
similar to water include hydrazine and DMF but these are somewhat less
available and considerably more dangerous.
Water also has the advantage of being non-flammable,
and noticeably inert to attack. The
preparation of water in a laboratory setting is simple and if it were not so
widely available it would be a easy task, the combustion of hydrogen in oxygen,
some acid base combinations react to produce a quantity of water, and thermal
decomposition of weak hydroxides. Water
is also relatively easy to purify from general inorganic contaminates, simply
by distillation. However when it comes
to organics a number of solvents form azeotropes with water that must be broken
before the two solvents can be effectively separated.
Tap water can work for most reactions but it is
better to used distilled water, it is available at most grocery stores and
pharmacies for a reasonable price.
Diethyl Ether CH3CH2OCH2CH3: [0.71 g/ml] The good: Ether is fairly inert, solvates a nice
variety of compounds, has a low boiling point (34.5 °C) and a very low freezing point (-116.2 °C) so you can drive it off completed reactions, ether was
used extensively in chemistry until recently so reactions using it can be
copied exactly, plus it can be made with relative ease. The bad:
Ether is highly flammable, it can form unstable peroxides in contact
with oxygen and if the ether is boiled down containing a high peroxide
concentration it can explode. Diethyl
ether is commonly referred to simply as ether or ethyl ether, if something
mentions using ether this is the ether it means [Note there is also a solvent
referred to as petroleum ether, this is not the same thing.]. The preparation of ether falls into the
advanced chemistry category, not in principle, but in practice:
CH3CH2OH
=[H2SO4 (Concentrated)]Þ CH3CH2OCH2CH3
+ H2O
So, ethanol heated with concentrated sulfuric acid
gives ether and water. The sulfuric
acid is necessary, not just a catalyst, its affinity for water is one of the
driving forces of this reaction, the ethanol is added to the hot acid, which is
well beyond the boiling point of the ethanol, the ether being distilled as it
is formed. The manufacture of ether at
home is a tradeoff with safety and privacy, ether is a somewhat watched
chemical in many places and ordering it form a supplier (assuming you find a
chemical supplier that is willing to sell it) may set up red flags which could
lead authorities to assume you are manufacturing illicit chemicals at your
home, but on the safety aspect, lacking the proper glassware this procedure is
exceptionally dangerous, even with the proper glassware there is a degree of
danger. Luckily there is usually a
substitutable solvent for ether depending on the reaction, still, it is a
useful solvent.
Acetone CH3COCH3: [0.79 g/ml] One good use for
acetone is the cleaning of labware. It
acts as an in-between solvent, grease that may not come off with water can be
pretreated with acetone to remove the bulk of the grease, then washed clean
with water, acetone being soluble in water and able to solvate many non-polar
molecules. It is also good for this
purpose for the reason that it has a high vapor pressure, once a piece of
glassware is washed with acetone the film left dries out quickly and the
glassware is ready for use. Acetone
also finds a place as a reactant, and solvent medium with a low melting point
of –94.3 °C and a boiling point of 56.2 °C it is more reactive then many other solvents though. It is flammable and subject to chlorination,
polymerization, and the haloform reaction among others. Preparation of acetone on a home scale can
be done from the pyrolysis of calcium acetate, but is not necessary, it is
available in many over the counter products for removing paint from fingernails
and additionally as a solvent in hardware stores, most of these products
labeled pure or 100% acetone.
Methyl-Ethyl ketone sold under the acronym MEK is another ketone
available on the market, it’s properties are similar to acetone and it will
also undergo the haloform reaction. It
is sold for stripping paint.
Methanol CH3OH: [0.79 g/ml] If something is
soluble in water, it is also usually soluble, to a lesser extent, in
methanol. From methanol formaldehyde
and formic acid can be made, and there are other reactions in which it can
readily participate. Methanol is
flammable but not incredibly so and is somewhat widely available for a number
of purposes, gas line defroster, hardware store solvent, and windshield washer
fluid. As a reaction medium it suffices
for some reactions, it has a boiling point of 64.5 °C and a freezing point of –97.8 °C, for many reactions though there are better mediums to
conduct them. The addition of methanol
or ethanol to saturated inorganic solutions in water usually results in the
precipitation of some or nearly all of the solvated salt. Overall though it is a good reagent to have
laying around. Consumption of methanol
can result in blindness and should be avoided.
It is not particularly hazardous as an inhalation hazard or contact
hazard but precautions should still be taken.
Ethanol CH3CH2OH: [0.82 g/ml] The properties of
ethanol are similar to those of methanol, the boiling point and freezing point
are shifted further up scale but other then that they behave closely to one
another. Ethanol and methanol are very
difficult to make anhydrous, ethanol forms an azeotrope with water that
contains a somewhat high percentage alcohol. But to go beyond that drying
agents / dehydrating agents start to become necessary. The prolonged action of anhydrous copper
sulfate on concentrated ethanol is one way to make a nearly anhydrous
product. Ethanol is available over the
counter for consumption in percentages up to 95% but it can be expensive from
this source, but the purity is somewhat guaranteed, it is additionally
available over the counter denatured as a painting supply, however the
denaturants can vary, ketones, methanol and other things can be added and other
impurities can be present since it is not intended for human consumption after
all.

Chloroform CHCl3: [1.5 g/ml] While technically
not a common solvent as it is not usually commercially available over the
counter, the preparation of chloroform is easy enough for the amateur chemist
and the reagents are easily acquired, a preparation is included in this text
(Under section 5). Chloroform is toxic
enough to where you should avoid unnecessary inhalation of the vapors and any
skin contact but is relatively safe overall. Chloroform was used for years as
the common solvent for organic material, the extraction of everything from
albumin to zein. Chloroform is slightly
soluble in water to the extent of about 8g/L but it still forms nice layers
when added to water.

Improperly stored
chloroform, notice the layer between the water and the bottom chloroform layer.
When stored usually 1% by volume ethyl or methyl
alcohol is added to retard its decomposition.
Chloroform normally decomposes to phosgene (COCl2, section 4.12), the
addition of these alcohols leads to the formation of the carbonate ester which
helps to slow decomposition. Chloroform
can be stored under water but doing so can result in additional decomposition,
even the oxygen from air can cause the decomposition of chloroform, but not at
a rapid rate. Chloroform has a liquid
range of nearly 100 °C,
from –63.5 °C to 61.2 °C, it was at one time used as an anesthetic but that use
was discontinued due to the toxic effects of chloroform. It is relatively inert in reactions, but
contact with solid hydroxides and strong oxidizing solutions should be avoided.
Methylene Chloride CH2Cl2: [1.3 g/ml] A surprisingly
useful solvent for extracting desired compounds in the liquid phase, it
possesses some unique solvent properties.
Chlorinate hydrocarbons are somewhat toxic though not excessively so,
still methylene chloride should be treated with respect. Methylene chloride is non-flammable, boiling
point of 40.1 °C
and a freezing point of –97 °C. The preparation of methylene chloride in a
home environment is only feasible by the chlorination of methyl chloride or
methane which is a quite dangerous, it is therefore fortunate that with some
searching methylene chloride can be procured from over the counter sources
specifically in paint thinners and removers, careful distillation from these
mediums can provide a product of suitable purity for most reactions.
Tetrachloroethylene CCl2CCl2: [1.60 g/ml] Available as a component of some paint strippers tetrachloroethylene is a
somewhat limited solvent. It also finds
use as a reactant, it has a boiling point of 121 °C and a freezing point of –22.4°C. As with all chlorinated hydrocarbons there
is a degree of toxicity to this compound and like most it is non-flammable.
Trichloroethylene CCl2CClH: [1.46 g/ml] It is imperative
the one never mix trichloroethylene with a strong base, doing so will likely
result in the formation of dichloroacetylene, a carcinogenic compound that
causes nerve damage. Trichloroethylene
is available over the counter in can form in auto part stores, under pressure,
for cleaning auto parts.

Toluene C6H5CH3: [0.87 g/ml] An aromatic
hydrocarbon, good all around non-polar solvent, that until recently had a wide
availability as a solvent for tough to remove paint. Reactive to halogens and other instances it is far from inert,
but suffices for reactions nonetheless.
Tolune has a boiling point of 110.7 °C and a freezing point of –94.5 °C.
Xylene C6H4(CH3)2: [0.86 g/ml] Properties similar to
toluene but has a wider availability still.
Xylene is also more reactive, the duel methyl groups activating it
further then the one on toluene. It is
a mixture of isomers, ortho, meta, and para xylene, and cannot be bought as
purely one component as xylene will exchange its methyl groups and within a few
days or weeks form a mixture of xylenes again.
Misc. Hydrocarbons: There are literally hundreds of hydrocarbons available on
the market to the amateur chemist. They
are used in many everyday applications most notably in nearly all combustible
fuels, gasoline, kerosene, diesel, and all other sort of combustible. Pure hydrocarbons are decent for removing
organics from an inorganic phase, but take special note of the additives that
may be added to these hydrocarbons to make their combustion more manageable,
especially in mixtures intended for the combustion engine.
Solubility Table:
M =
Miscible (soluble in all proportions) Ss = Slightly
Soluble / Somewhat Soluble Is = Insoluble
|
----- |
H2O |
CH3OH |
Acetone |
Ether |
Ethanol |
CHCl3 |
CH2Cl2 |
CCl2CCl2 |
CCl2CClH |
Toluene |
Xylene |
|
H2O |
----- |
M |
M |
Ss |
M |
Ss |
Ss |
Is |
Ss |
Is |
Is |
|
CH3OH |
M |
----- |
M |
M |
M |
|
|
|
|
|
|
|
Acetone |
M |
M |
----- |
M |
M |
M |
|
|
|
|
|
|
Ether |
Ss |
M |
|
----- |
M |
M |
|
|
|
M |
M |
|
Ethanol |
M |
M |
M |
M |
----- |
M |
|
|
|
|
|
|
CHCl3 |
Ss |
M |
|
M |
M |
----- |
|
|
|
M |
M |
|
CH2Cl2 |
Ss |
M |
|
M |
M |
|
----- |
|
|
|
|
|
CCl2CCl2 |
Is |
M |
|
M |
M |
|
|
----- |
|
|
|
|
CCl2CClH |
Ss |
|
|
|
|
|
|
|
----- |
|
|
|
Toluene |
Is |
|
|
|
|
|
|
|
|
----- |
M |
|
Xylene |
Is |
|
|
|
|
|
|
|
|
M |
----- |
Purifying Solvents:
Not
a necessary step if your solvents are purchased from a lab supply. However if you purchase your solvent over
the counter and it was not being marketed with a purity somewhere on the bottle
you can not trust it entirely. As a
matter of fact there is a suspicion that should be associated with many of the
chemicals your purchase over the counter unless it is something that explicitly
states the purity or is meant for human consumption as a pure product (e.g. citric
acid). Even if you make a solvent on
your own additional purification is usually necessary, for instance, chloroform
produced via the haloform reaction is often contaminated with water,
insolubles, and acetone for starters.
Ether produced from ethanol and sulfuric acid often contains unreacted
ethanol, water, and sulfuric acid/sulfur dioxide contamination. So in these cases there also exists a degree
of contamination that must be accounted for.
The
normal first line of defense for a solvent is to shake with a drying agent,
decant, and distill. Non-volatile
products such as anything dissolved in your solvent will not carry over and by
using a fractioning column and paying careful attention to the temperature of
the distillate purity can be increased significantly in one run. There is one danger here though, aside from
the inherent flammability of most solvents, peroxides may be present in your
solvent, either from impurities or in the case of ethers, the solvent
itself. Therefore it is helpful to
shake with some kind of reducing agent initially or to simply discontinue
distillation with a noticeable amount of liquid left in the distilling flask
(due to your intention to separate impurities one should not distill to dryness
anyway, not that distilling to dryness should be done in any other
situation).
Mixing Solvents:
Mixing solvents of different properties to give a solvent
system of a desired property is to some extent hit and miss to the amateur
chemist. But mixed solvents do have their definite advantages. They can cause the precipitation of an
undesirable compound while keeping your desired compound in solution and vice
versa. Additionally they can simultaneously
put two compounds in solution to react that might otherwise be in different
phases. Some solvents also have
catalytic properties on a reaction and their inclusion in the reaction medium
might speed things up greatly. Despite
these great positive aspects mixed solvents are often overlooked because
despite some solvent system looking good on paper (and other solvent systems
having nearly impossible to predict properties) many of them just don’t give
the expected results upon actually trying the experiment. Which results in wasted solvent, wasted
time, and wasted reagents. None the
less they do come into play on occasion, the following are some examples.
-
Dissolving (Somewhat) Nobel Metals:
Although many metals will simply dissolve, given enough time in hydrochloric acid or sulfuric acid or even acetic acid, there are some, which won’t care in the slightest about being put into these environments. Although not strictly limited to the following, here are some examples of metals that may require a bit of special treatment to put into solution: (1) Silver; (2) Copper; (3) Bismuth; (4) Nickel; (5) Mercury
Although we are focusing on metals like those just listed, there are many other metals with which you would find difficulty putting into solution, like tungsten, tantalum, and in their own category gold and the platinum metals (platinum, iridium, rhodium, and a few others.), which have their own difficulty, associated with their dissolution. Here though are presented a few methods that may aid in dissolving the metal of your choice to form a solution of suitable cations.
Displacement- The principle here is simple, you find a readily available salt containing a metal cation that has a high reduction potential such as copper sulfate (Cu2+(aq) + 2e- Þ Cu(s) V=.52) and add to it solid pieces of the metal whose salt you desire, such as lead (Pb(s) Þ Pb2+(aq) + 2e- V=.13) The net reaction, lead going into solution is favored by the difference in voltage between the two. This particular reaction is complicated by the formation of lead sulfate on the surface of the lead reacting which only has a limited solubility, to rectify this a fish aquarium bubbler could be led into the area where the lead is reacting, the agitating action of the bubbles continually removing the sulfate layer and deposited copper from the lead allowing it to react further. Of course this is not good for putting metals into solution with very high potentials such as
Electrolysis- As electrolysis of a solution proceeds reduction occurs at the cathode and oxidation at the anode. In a solution containing only a little electrolyte such as NaCl with nickel electrodes the usual reaction is the formation of hydrogen and oxygen, if the concentration of NaCl is increased some chlorine my form in place of oxygen, however if in place of water a strong acid solution is used, such as HCl(aq) and the electrolysis preformed with sufficient current and voltage a significant portion of your cathode may be quickly reduced and put into solution, in this experiment the relevant equation being:
Ni(s) Þ Ni2+(aq) + 2e-
Products at the anode include oxygen and chlorine gas, hydrogen is also produced at the cathode from reaction with finely divided nickel and such, but the overall effect in this case would be the quick production of nickel chloride, and by coiling your cathode to increase the cathode surface in the electrolyte and decreasing the surface area of your anode in the solution you can very efficiently make a concentrated solution of many cations, evaporation in the case of hydrochloric acid would volatize off remaining acid leaving a somewhat pure product.
Mixtures of acids with oxidizing agents-
Fusion with hydroxides-
Specialty acid combinations-
 4.10
Halogens
4.10
Halogens
Pictured at left is a wonderful example of one of the trends followed by the halogens. As you move down the group the oxidizing power of each element decreases. So it stands that the halogen preceeding each halogen should have the oxidizing power to oxidize the element beneath it and in this way replace it. In this instance chlorine is being generated in the flask on the left and goes up through a piece of glass tubing and into a gas washing bottle. The bottle contains a concentrated solution of sodium bromide and as the chlorine goes through it a simple reaction takes place:
Cl2(g) + 2NaBr(aq) Þ 2NaCl(aq) + Br2(aq)
The bromine thus generated being slightly soluble in water and coloring the solution red to indicate its presence. It follows that chlorine would be able to easily do this to iodide in solution and that bromine would also be able to do this to iodine but no to chlorine. These three halogens also have other predictable trends, they tend to form soluble compounds with the exception of their silver salts and their copper (I) salts and they each have a series of oxoacids which are similar to one another. Because of these similarities in some reactions halogens are generically represented with an X, for example CH3X would stand for either methyl chloride, methyl iodide, methyl bromide, or less likely methyl fluoride. When written in reactions and such the halogens are written F2, Cl2, Br2, and I2, the term for this being diatomic, which means the halogens do not go around as free atoms but rather each halogen is bonded to a neighbor, in this case by a single bond, e.g., Cl-Cl the bonds however are somewhat weak and UV light will usually do the trick to rupture these bonds and leave a reactive radical containing an unpaired electron, the strength of this bond is weakest with fluorine and strongest with iodine. Each of these halogens also have a whole series of oxidation states although they are not the same for each halogen they include –1, +1, +3, +5, and +7 oxidation states.
|
|
HX |
HOX |
HOXO |
HOXO2 |
HOXO3 |
|
Fluorine |
Hydrofluoric Acid |
Hypofluoric Acid |
DNE |
DNE |
DNE |
|
Chlorine |
Hydrochloric Acid |
Hypochloric Acid |
Hypochlorous Acid |
Chloric Acid |
Perchloric Acid |
|
Bromine |
Hydrobromic Acid |
Hypobromic Acid |
Hypobromous Acid |
Bromic Acid |
Perbromic Acid |
|
Iodine |
Hydroiodic Acid |
Hypoiodic Acid |
Hypoiodous Acid |
Iodic Acid |
Periodic Acid |
But what about fluorine? Fluorine is the odd element out, it does not follow these trends. Fluorine has one major oxidation state –1 there are few compounds where this comes into question but those instances are few and far between. Fluorine is the most electronegative element in the periodic table and will react with most elements at room temperature. Whereas the other elements have series of oxoacids of the form HOX, HOXO, HOXO2, and HOXO3 where X is the halogen in question fluorine only displays hypofluoric acid fleetingly by passing flurorine over moist glass at low temperatures. Above this fluorine would be in a higher oxidation state and as far as modern chemistry can show these forms do not exist.
Really though chlorine is the best example of this series, all of the oxoacids having a ‘simple’ method of preparation, chlorites (salts of hypochlorous acid) and hypochlorites (salts of hypochloric acid) being the most unstable of the series. Whereas perbromic acid is only prepared with great difficulty by passing fluorine through a basified bromate solution and iodine compounds of the form HOI and HOIO being preparable only in dilute solutions, periodic acid also displaying some deviance from the remainder of the family by having a formula of H5IO6, two water molecules having found their way into the molecule and sticking there pretty good.
Aside from being the basis of many of the most prevalent acids in chemistry the halogens also serve to offer a diverse group of oxidizing agents with fluorine and iodine at the extremes and the oxoacids forming an even larger middle. Many halogen salts are fairly soluble in aqueous inviorments and are somewhat stable to oxidation and reduction. Another shining point of the halogens comes in their organic reactivity, reacting directly with alkanes to give hydrogen halides and substituted alkanes (halogenated hydrocarbons) which are great starting points in organic synthesis. Really the halogens comprise the most utilitarian family in the periodic table and their use in the lab should not be underestimated.
Fluorine
 Chlorine
Chlorine
Molecular Weight 35.45
Slightly soluble in water, soluble in non-polar solvents
Green/Yellow dense gas
-1, +1, +3, +5, +7 Oxidation States
Chlorine is somewhat
simple to prepare and there are a number of methods to do so, even from over
the counter products. Just as with
bromine the pool industry is a great help in the preparation of chlorine,
simple acidification of many pool chlorinators such as sodium hypochlorite (a
liquid chlorinator that is also the active ingredient in bleach), calcium hypochlorite
(a powder sold in packs often under the name “Shock”) and trichloroisocyanuric
acid and trichlorotriazinetrione (yet more pool chlorinators) can be instant
sources of this highly noxious gas.
Additionally it can be prepared by electrolysis of concentrated aqueous
salt solutions with relatively high current densities, as well as by the
oxidation of chlorine in the –1 state such as in hydrochloric acid, usually
with permanganate. These methods will
all give ‘wet’ chlorine of some purity or another that should be dried before
use, chlorine being usually dried by passing through concentrated sulfuric
acid.
 Bromine Br2
Bromine Br2
Molecular Weight 79.904 g/mol
Slightly soluble in water, soluble in non-polar solvents.
Red vapor, liquid can be nearly black when present in large amounts.
-1, +1, +3, +5, +7* Oxidation States (* = very difficult to
prepare)
One of bromine’s claim to fame is of course that it is only one of two common elements that are liquids at room temperature and pressure. Because it is a liquid it is also somewhat easily storable by the amateur chemist, making it the strongest of the readily storable halogens. Bromine finds a wide variety of uses in the home lab. It can create a large number of bromides by direct reaction with elements, pictured below is the reaction between bromine and aluminum turnings:
2Al(s) + 3Br2(l) Þ 2AlBr3(s)

In this case the reaction has a special utility, whereas aluminum bromide could also be prepared from hydrobromic acid and aluminum, the hydrate is formed and the product is nearly impossible to dehydrate. The anhydrous form is nearly the only good form of aluminum bromide to use in organic chemistry, and as such straight bromine dried by shaking with sulfuric acid is one viable way to produce this useful compound. There are many ways to produce bromine, but the most reasonable methods of course use the most readily available materials. Thankfully bromine compounds have found some utility in the pool and spa industry and as such the most readily accessible sources of bromine involve either sodium bromide or in the form of complex organic compounds that hydrolyze in water to give hypobromous acid (HOBr) such as tribromo-s-triazinetrione, treatment with acid will convert these readily to the bromide anion and put you in the same predicament as you would have been had you used NaBr from the beginning.
When going from sodium bromide to free bromine there is really only one key ingredient, an oxidizing agent under acidic conditions. If the reaction were under basic conditions then the bromine would react immediately to from hyprobromite and bromates. The acidic environment also helps the oxidizing agent do its job. Nearly any oxidizing agent will work, peroxide, manganese dioxide, hypochlorite, permanganate, even oxygen bubbled through an acidic solution of bromine can work were it not for the bubbling of gasses going though the liquid sweeping away the bromine as it were formed. Note however bromines low boiling point of 59 °C. But even well below this bromine will readily volatize off and fill an area with its choking fumes. As such preparations of bromine should have ice added, and take place as cold as feasible to prevent it from filling the area and reducing yields.
 Disposing
of bromine either dissolve in water or on its own is simple. Just adding the solution to an aqueous
solution of a base will do the job readily (see picture at right). However the base must be completely
dissolved, if you attempt to add bromine water to a hydroxide producing agent
recently tossed into some water you will not get nearly the same
reactivity. Always wear gloves when
working with bromine as its contact with skin will discolor it and leave painful
lingering sores which are slow to heal.
A reducing agent such as citric acid solutions or sodium thiosulfate
solutions can be used to treat recent areas of contact with bromine.
Disposing
of bromine either dissolve in water or on its own is simple. Just adding the solution to an aqueous
solution of a base will do the job readily (see picture at right). However the base must be completely
dissolved, if you attempt to add bromine water to a hydroxide producing agent
recently tossed into some water you will not get nearly the same
reactivity. Always wear gloves when
working with bromine as its contact with skin will discolor it and leave painful
lingering sores which are slow to heal.
A reducing agent such as citric acid solutions or sodium thiosulfate
solutions can be used to treat recent areas of contact with bromine.
Bromine attacks most every metal including many from the platinum group. But dry bromine can have a hard time of attacking some other metals, notably some metals that would seem quite reactive such as magnesium, lead, iron, zinc, and even sodium. Aside from these metal compounds bromine will attack non-metals such as sulfur and also forms a large series of interhalogen compounds, interesting examples being bromine triflouride and bromine pentaflouride, the pentaflouride exploding on contact with water. Bromine chloride finds more use as a chemical intermediate oxidizing agent. Bromine itself is a good oxidizing agent, in inorganic preparations it has powerful oxidizing properties by adding to hot solutions of hydroxide, in this manner ferrates and bismuthate can be produced. In organic preparations bromine is less reactive then chlorine and is thus more selective in brominations. Brominated organic compounds being of great utility in synthesis operations.
On distillation from an aqueous solution bromine carries over roughly 2% of its weight in water. This is not a real azeotrope but it is something to be noted. Normally bromine can be generated whereupon it sinks to the bottom of the solution it was generated in where it can be pipetted off from. This can be used directly for some things but further purification can be achieved through non-distilative measures. Normally by first washing with a small amount of water, then shaking with concentrated sulfuric acid and finally by filtering though glass wool and storing under water or sulfuric acid.
Iodine
Not only are the alkali metals interesting for their reactivates and unusual properties, they exhibit definite trends as you move down the period which are easy to memorize.
· Lithium is the hardest alkali metal, but it can still be cut with a knife, as you move down the period from lithium to cesium the metals get softer and their melting points go down as well, cesium is a liquid only slightly above room temperature.
· Lithium is the least reactive of the metals, cesium the most, francium is only present on the earth in gram amounts at any time but it likely continues this trend.
· All alkali metals exhibit the +1 oxidation state as their main and only common oxidation state.
· Atomic radius increases as you move down the period, cesium, the largest stable element in the periodic table can stabilize the triodide anion I3- due to its size.
· The increasing reactivity of the alkali metals can be seen in their oxide formation, lithium forms predominately the normal oxide Li2O, sodium mainly the peroxide Na2O2, potassium mainly the superoxide KO2, and rubidium and cesium form almost entirely the superoxide. Additionally they form somewhat stable ozides CsO3 by passing ozone over their hydroxide and separating the formed ozide from remaining hydroxide by solubility differences in liquid ammonia.
· All are soluble in liquid ammonia or hydrazine yielding brilliant blue solutions that contain 'solvated electrons'. These are powerful reducing agents.
· The alkali metals react with water along the lines of M(s) + H2O(l) ---> MOH(aq) + 1/2 H2(g) Lithium is somewhat manageable, sodium will usually ignite and can ignite clouds of hydrogen above it leading to explosion, by the time you get to cesium it will detonate.
· Lithium is the least dense metal of the period, it will float on most any oil you try to protect it under, cesium is the most dense.
Distinguishing between the different alkali metals in solution can be incredibly difficult chemically as they all behave very similarly and most salts are very soluble. The easiest test to distinguish between the alkali metal cations in solution is the simple flame test. A circle of wire, preferably inert, e.g. platinum, is dipped into a concentrated solution of the salt. It is then put into a high flame and the color of the flame observed.
Lithium = Red
Sodium = Orange/Yellow
Potassium = Purple
Rubidium = Red - Violet
Cesium = Blue
However the tests can be easily false, the colors can be over-run by other colors generated, and sodium, the most common contaminate of the other alkali metal salts due to most of them being produced from it, can easily over-shadow the other more sensitive colors of potassium and the like, leading to a false positive for sodium.
Lithium

Lithium is very light, it will actually float in oil.
Like other elements that start off their group lithium shows some properties that differ from the normal properties of the rest of the table. When exposed to air lithium will form a black coating of nitride Li3N which reacts with water to produce ammonia. One of the only other metals that will do this is magnesium metal appropriately heated, which similarly forms the nitride. Nearly all lithium salts are hydrated and removing the waters of hydration are nearly impossible on some of them, particularly the chlorate and perchlorate that decompose before a majority of the water has been removed. Lithium perchlorate actually contains more oxygen, on a volume-to-volume basis, then liquid oxygen. The hydroxide of lithium forms a stable hydrate LiOH*8H2O and does not dissolve in water if left exposed to the atmosphere, but it will form the carbonate like the other alkali metals, reacting with atmospheric CO2. The carbonate of lithium is the least stable carbonate of the alkali metals and decomposes around 1000C.
Lithium metal has a high electrode potential and therefore it is becoming popular in some batteries (this also means that it can be very reactive in the liquid state, it will destroy glassware when liquid). Lithium is one of only a handful of metals that have actually become more expensive over the last 20 years. Lithium is usually stored on top of oil under an argon atmosphere.
Sodium

This is the most common alkali metal that we run across. Sodium carbonate, sodium chloride, sodium vapor lights, sodium hydroxide, sodium bicarbonate, the list goes on. Sodium is the most abundant of the alkali metals in the earths crust and it shows in our preference to it, that and the fact that we need sodium chloride in our daily dietary intake.
Potassium
 Potassium
follows the trend set up between sodium and lithium in that it is more reactive
then either of them, being further down the group. Therefore when it
burns in air, shown left (Notice the purple flame that is also indicative of
potassium ions in a flame test), it produces not only the oxide and peroxide,
but predominately the superoxide. KO2 is a very powerful oxidizing agent
and it proves to be a nuisance when storing potassium. When a block of
potassium is stored under mineral oil or another inert substance unless it is
in an air tight container and the liquid has been degassed the potassium can
pick up oxygen and form the superoxide. This leaves a yellow-orange
coating on the surface of the pieces of potassium. This in and of itself
is usually no problem however upon cutting into a piece of potassium covered in
this coating it can force the superoxide into the unreacted potassium and in
worst cases this can cause an explosion, sending flaming bits of potassium
everywhere and causing severe damage to the individual performing the
manipulation.
Potassium
follows the trend set up between sodium and lithium in that it is more reactive
then either of them, being further down the group. Therefore when it
burns in air, shown left (Notice the purple flame that is also indicative of
potassium ions in a flame test), it produces not only the oxide and peroxide,
but predominately the superoxide. KO2 is a very powerful oxidizing agent
and it proves to be a nuisance when storing potassium. When a block of
potassium is stored under mineral oil or another inert substance unless it is
in an air tight container and the liquid has been degassed the potassium can
pick up oxygen and form the superoxide. This leaves a yellow-orange
coating on the surface of the pieces of potassium. This in and of itself
is usually no problem however upon cutting into a piece of potassium covered in
this coating it can force the superoxide into the unreacted potassium and in
worst cases this can cause an explosion, sending flaming bits of potassium
everywhere and causing severe damage to the individual performing the
manipulation.
Potassium superoxide and the superoxides of other higher alkali metals react with water along the following equation:
2KO2(s) + 2H2O(l) ---> 2KOH(aq) + H2O2(aq) + O2(g)
|
Potassium superoxide is also used in space capsules for the duel purpose of sequestering CO2 from the astronauts breath and to generate additional oxygen. It is also used in some self contained breathing apparatuses: 4KO2(s) + 2CO2(g) ---> 2K2CO3(s)
+ 3O2(g) |
Another interesting property of potassium is that it forms a carbonyl compound K(CO)6 however it is not the most stable of carbonyl compounds by a long shot. It can explode for no reason at all at STP therefore any reaction that generates elemental potassium, or uses elemental potassium, in the presence of carbon monoxide (i.e. reduction of the carbonate with charcoal) should be treated with caution as the in situ preparation of potassium carbonyl may cause explosions.
Potassium-sodium alloys are liquids at STP and more reactive then either metal individually. Industrially potassium is prepared by distilling it from a mixture of sodium metal and potassium chloride. The replacement of potassium with sodium in the reaction is not immediately sensible due to potassium being the more reactive, however the reaction works due to the potassium formed having a significantly lower boiling point then the sodium therefore the reaction is pushed foreword as the potassium boils off. The Castner cell also works with potassium hydroxide in place of sodium hydroxide and actually gives better yields and a lower melting solid. However potassium is more flammable and reactive therefore this reaction is less favored. The electrolysis of the chloride is also less favored due to higher working temperatures of the eutectic. Thermite type reactions also work for the production of potassium, reducing potassium oxide or hydroxide with magnesium works, but aluminum forms aluminates that decrease yields significantly. The most common potassium salt available to the amateur chemist is potassium chloride, it is widely available for use in water softeners and as a salt substitute in health food areas.
Rubidium / Cesium
4.12 Functional Groups of Organic Chemistry
4.13 Gasses
 Working with gasses can be quite
important to a chemist. They not only
posses a great variety of reactivity, but many of them are also easy to
make. Dealing with gasses is of course
different then working with liquids or solids but the change over is easy as
long as you take it step by step. Your
main concern is actually probably going to be how the flow of gasses though
your system is going to affect fluids in your system, and how those fluids will
respond if pressure is applied in the opposite direction if for some reason gas
generation slacks off or stops. By
nature gasses can be everywhere around you, and unlike a solid or liquid which
has to first act on the skin before causing ill effects gasses can go straight
from the air into the blood stream.
Therefore gasses possess and added degree of possible danger. All gasses are asphyxiants in large amounts
(except oxygen) but many possess additional hazards. The author of this text recommends against working with several
gasses based on the extreme toxicity of those compounds. However there are still a number of other
gasses that can be safely worked with and handled. Please treat all gasses with respect and plan out your reactions
and apparatuses before hand and you will be rewarded with safety.
Working with gasses can be quite
important to a chemist. They not only
posses a great variety of reactivity, but many of them are also easy to
make. Dealing with gasses is of course
different then working with liquids or solids but the change over is easy as
long as you take it step by step. Your
main concern is actually probably going to be how the flow of gasses though
your system is going to affect fluids in your system, and how those fluids will
respond if pressure is applied in the opposite direction if for some reason gas
generation slacks off or stops. By
nature gasses can be everywhere around you, and unlike a solid or liquid which
has to first act on the skin before causing ill effects gasses can go straight
from the air into the blood stream.
Therefore gasses possess and added degree of possible danger. All gasses are asphyxiants in large amounts
(except oxygen) but many possess additional hazards. The author of this text recommends against working with several
gasses based on the extreme toxicity of those compounds. However there are still a number of other
gasses that can be safely worked with and handled. Please treat all gasses with respect and plan out your reactions
and apparatuses before hand and you will be rewarded with safety.
A gas bubbler, as show on the left can be quite the useful tool for an at home lab. However the construction of such a device is exceedingly simple and therefore purchasing a piece of equipment like the one shown can be avoided. The basic principle is simple, gasses come in through the tube on the left, bubble though a solution contained in the body of it, going though a glass frit on the way to disperse them better thereby creating more surface area causing the gasses to be absorbed/washed better, and finally exit though the tube on the right which comes nowhere near the water thereby preventing the liquid in the container from being transferred to the next container.
If you expect to be working
with gasses a lot you might want to ask yourself if you want to create a fume
hood. Working in a fume hood is like
working in a box made of glass with only one exit for any gasses, though a tube
away from you. Usually drawn though the
tube with a fan and treated to neutralize them, fume hoods are indispensable
for some, but not many, applications.
Gas generation and handling setups:
Above is a very simple gas generation apparatus. However since it lacks a trap (which would prevent fluid from coming back into the flask) it would only be good for low temperature generations, which would prevent a dangerous suckback. Something where the addition of the liquid in the sepretory funnel would reliably keep the gas coming along and where your intention is to dissolve your gas in a liquid in which it is fairly soluble (because anything that doesn’t dissolve is just going to get into the air around you).
This apparatus goes two steps further, one, it incorporates a trap to
prevent liquid from flowing back into the possibly hot flask that may or may
not react with water, and two, it has a final gas scrub which may possess a different liquid then
the original absorbent flask to make sure to destroy harmful vapors.
Even one step further, this setup has an initial trap, then a wash to take out impurities that may hurt the reaction, followed by another trap and finally the absorbance and the scrubbing step. Also there is a separatory funnel for the addition of another liquid to the reaction flask to keep the reaction going.
Tips for working with gasses:
· When working with flammable gasses it is necessary to heat any part of your vessel with a non-flame, non-sparking heat source. The smallest spark can trigger an explosion and special care should be taken to remove sources of ignition from your work area.
· In the case of gasses that are highly unstable (arsine, diborane, phosphine, silane, hydrazine) and can decompose exothermically when heat is applied and if it is a case where that gas must be disposed of by incineration, it may be necessary to run the gas into a container containing damp sand, up though the bottom and though a tube at the top, this will negate the possibility of a sudden explosion of gasses flashing back and detonating your whole reaction setup.
· If working with a gas of very high toxicity (most of them on the list) and inhalation occurs, do not delay, get medical help immediately, gasses are absorbed fast and a delay of a few minutes could mean your life, call a poison control, or the local emergency services.
· Make sure your reaction apparatus is air tight beforehand, and keep some duct tape around for emergency fixes (real emergencies only, if some of these start to leak you might just evacuate before it gets to the duct tape)
· Flammable/Explosive gasses should never be generated in the same reaction as one that also produces oxygen or nitrous oxide, the possibility of explosion is too great.
Gas Masks:
|
Gas |
Filter Properties |
|
Hydrogen Sulfide H2S |
Filters designed specifically to protect against hydrogen sulfide can only do so for very limited periods of time and are designated rescue filters. Being that they have a life of less then 10 minutes, are not designed for excessive concentrations, and are expensive, they are not economically feasible. Hydrogen sulfide is extremely toxic and has killed many individuals working in amateur labs because it quickly deadens the sense of smell. Common logic would dictate that you just not work with this foul smelling gas. |
|
Organic Solvents |
There are filters specifically designed to block out VOC (Volatile Organic Carbons) these work fairly well and are the most commonly available filters in my experience as they are used widely in painting. They last for an extended period of time and are designed for constant use. Good for working with solvents of all kinds but especially carcinogenic solvents such as halogenated hydrocarbons and benzene derivatives. (Methanol is notoriously difficult to filter out, however it does not present much of an inhalation hazard.) |
|
Hydrogen Chloride Chlorine Acid Gasses |
There is a specific filter known as an acid gas filter. It will block out hydrogen halides (except fluorides) and elemental halogens along with other acidic gasses such as SO2, although they cannot reliably block out nitrogen oxides. |
|
Hydrogen Cyanide HCN |
Regular carbon filters can block out HCN, however one exposure can ruin the filter and therefore make you susceptible to anything else you attempt to protect yourself from. As with H2S, it is better not to use this gas/liquid in your experimentation, as it is highly toxic. |
To put on a gas mask you place it
over your face, straps behind your head, the exact technique is not as
important as the tests you do before giving it the all clear. First is
the positive pressure test, find the exit hold for the air, usually right in
the middle of the mask, cover it with your hand and breath out, if the air only
'farts' out from around the edges of the mask you are good. Next is the
negative pressure test, cover the intakes on your cylinders with your hands and
inhale, no air should come in, the mask should attempt to deform inward to
compensate for the uptake of air. If you pass both of these tests your
mask is correctly positioned on your face. If either one of these tests
prove negative, reposition your mask and try again.
Dealing with exit gasses:
In all instances read the below information
and thoroughly acquaint yourself with a gas before working with it. You are responsible for your own actions and
therefore should take extra precautions, gasses are inherently dangerous so use
them with care, extra information regarding the scrubbing of gasses is in the
information of the main text regarding specific gasses.
|
Name of Gas: |
Disposal by incineration: |
Disposal by scrubbing |
|
Acetylene |
Yes |
Possible (Oxidizing Agents) |
|
Air |
NA (Vent) |
NA (Vent) |
|
Ammonia |
Yes |
Yes (Acids) |
|
Arsine |
Not Recommended |
Yes (Oxidizing Agents) |
|
Butane |
Yes |
No |
|
Carbon Dioxide |
NA (Vent) |
Possible (Bases) |
|
Carbon Monoxide |
Yes |
No |
|
Carbonyl Chloride |
No |
Yes (Hot Aqueous Bases) |
|
Chlorine |
No |
Yes (Bases) |
|
Chlorine Dioxide |
Could Cause Explosion |
Yes (Bases) |
|
Boranes |
Yes |
Yes (Basic Oxidizing Agents) |
|
Ethylene |
Yes |
Yes (Oxidizing Agents) |
|
Fluorine |
No |
Yes (Anything) |
|
Hydrogen |
Yes |
No |
|
Hydrogen Cyanide |
Not Recommended |
Yes (Bases) |
|
Hydrogen Fluoride |
No |
Yes (Bases) |
|
Hydrogen Halides |
No |
Yes (Bases) |
|
Hydrogen Sulfide |
Yes (But Generates SO2) |
Yes (Bases/Oxidizing Agents) |
|
Methane |
Yes |
No |
|
Nitric Oxide |
Not Recommended |
Yes (Bases) |
|
Nitrogen |
NA (Vent) |
NA (Vent) |
|
Nitrogen Monoxide |
No |
Yes (Strong Bases) |
|
Nitrous Oxide |
Yes |
No |
|
Nobel Gasses |
NA (Vent) |
NA (Vent) |
|
Oxygen |
NA (Vent) |
NA (Vent) |
|
Ozone |
Not Recommended |
Yes (Reducing Agents) |
|
Phosphine |
Yes |
Yes (Oxidizing Agents) |
|
Propane |
Yes |
No |
|
Silane |
Yes |
Yes (Oxidizing Agents) |
|
Solvent Vapors |
Yes |
No |
|
Sulfur Dioxide |
No |
Yes (Bases) |
Scrubbing Exit Gasses: When scrubbing exit gasses care should be taken to ensure complete neutralization of the toxic effects. The greater the toxic effect the more drastic measures should be taken. Severely toxic chemicals should go though no less then two scrubbing solutions, but preferably three or more. The concentration of the solution should not be incredibly high or low but will depend on the gas to be neutralized. If it will help the addition of an acid base indicator may tell when some acidic or basic gasses have used up the neutralization power of a given solution.
Igniting/Incinerating exit gasses: When igniting exit gasses it is good to flush the system with an inert gas first (carbon dioxide, butane, propane, methane, argon, etc.) even steam will work for this purpose. This is so once flammable vapors come over and they are ignited the vessel does not contain any oxygen gas which may flash back into the system and blow up your glassware. If flushing the system is not feasible, let the apparatus run and generate gas, then from a distance blow away the gasses with a fan and ignite after the vessel is full of the gas in question (this method is not advised). Sometimes the gasses will escape the vessel at a speed sufficient to maintain a constant flame. However it is best to run the gasses though a tube that connects to a pipette and run them straight into a flame or into the intake of a flame. In this way if the gas evolution slacks off and the escaping gasses stop combusting but start exiting again afterwards they will still be burning as they exit. Also even spontaneously flammable gasses are not spontaneously flammable in all concentrations, without external burning some will make it all the way to the inside of your lungs and wreak havoc.
Specific Gasses: (Green
= Water Solution Basic Red = Water Solution Acidic) WS = Water Soluble
Acetylene HCCH: The most common source of
acetylene for the home chemist is by the action of water on calcium
carbide.
2H2O(l)
+ CaC2(s) Þ HCCH(g) + Ca(OH)2(s/aq)
Acetylene is quite the unsaturated molecule, being
that it contains a triple bond between the carbons. This can be acted on by a number of reagents, most of the halo
acids adding across it forming vinyl halides and it is easily oxidized or
polymerized. Acetylene is ridiculously
explosive if allowed to accumulate in one area. If liquefied it can undergo hazardous polymerization and as such
cylinders of acetylene sold for welding actually contain acetylene dissolved in
acetone. The cylinders of MAPP gas
available from hardware stores contain an derivative of acetylene among other
things. Acetylene has the normal
asphyxiation hazard that many of the gasses here have. It is more reactive then your average
hydrocarbon and is readily produced as stated before, calcium carbide still
being somewhat easily available specifically for the purpose of making acetylene,
especially over the internet. Acetylene
should not be lead into basified solutions or acidified, doing so can make
explosive acetylides or carcinogenic vinyl halides. Acetylene can only really be disposed of by incineration and then
the vessel from which the acetylene comes from must be purged of oxygen
otherwise the flame could flash back inside and detonate the vessel
Air: Air is a mixture of gasses of approximate composition by
volume; (78%) Nitrogen, (21%) Oxygen, (1%) Argon, (<1%) Other gasses, CO2,
Ne, He, etc. [Exact composition of the
air around you varies with your elevation and your surroundings, however these
numbers are relatively constant] Unless you take steps to the contrary, you
will be working in air. Most reactions
can be carried out with exposure to the atmosphere, there are many that cannot
be though, be sure to take into consideration the properties of the atmosphere
you make for your reaction, and how it will react with your reaction before
beginning any involved chemical reaction.
Never forget that the water content varies daily and that it can impact
many reactions to a great extent.
Ammonia (NH3) WS: Ammonia is not only
a foul smelling gas, but also a low temperature reaction solvent. It does the most interesting trick often
referred to as the “Solvated Electron” trick in which it will dissolve the
alkali metals which turns the ammonia a beautiful blue color and upon
concentration makes the ammonia look golden like liquid metal. Still though, these reactions take place in
liquid ammonia, so below –33 °C. Ammonia can be liquefied at home but it is a
hassle. Ammonia gas in the presence of
water attacks a number of metals but most notably copper with which it forms a
dark blue complex, note that ammonia is flammable and can be explosive if
initiated like the lighter hydrocarbons.
Ammonia gas is poisonous and its basicity does not combine well with
ones eyes and it causes terrible burning and tearing and eventually blindness. Commercial ammonia solutions available from
grocery stores and such contain roughly 4% ammonia by volume. Solutions of ammonia in water are also
referred to as ammonium hydroxide due to the equilibrium:
NH3(aq)
+ H2O(l) Û
NH4OH(aq)
However this is an equilibrium reaction and the
equilibrium actually lies to the left.
Ammonium hydroxide itself is not isolatable in the pure state, it is
known through its salts and in solution.
Ammonia is a very useful reagent to have around. It forms numerous complexes and has a wide
variety of reactions to exploit. Strong
solutions of ammonia can be made with the help of an ammonia salt, in this case
ammonium sulfate:
(NH4)2SO4(aq)
+ 2NaOH(aq) Þ
2NH3(g) + Na2SO4(aq) + 2H2O(l)
The combination of a solution of ammonium sulfate
and sodium hydroxide giving off noticeable ammonia gas readily. This reaction can also be done in the solid
phase but it can become violent. The
gasses being produced in both instances being channeled through water to
dissolve as much as possible then scrubbed with a solution of nearly any acid,
but the stronger the better.
Arsine (AsH3) WS: Arsine has a
garlic-like odor similar to phosphine.
Stibine (SbH3) is similar to arsine and this may be used as a
guide for it as well. Initial exposure
to arsine produces few symptoms, headache, nausea, nothing to make a person
worry too severely. However several
hours or so after what some would call a mild exposure, a breath or two,
vomiting and cramping set in and depending on the dose kidney failure, CNS
depression, and death. Arsine is the
most toxic way for the body to come into contact with arsenic. Because of its severe toxicity arsine should
be avoided. To destroy arsine from exit
gasses it is prudent to run the gasses though at least two washes of sodium
hypochlorite, calcium hypochlorite, potassium permanganate, bromine water, or
sodium hypobromite solutions. Be sure
their volume is sufficient to provide excessive decomposition ability for more
arsine then you can think might be produced.
Do not run arsine into any incineration, fine As2O3
will be produced creating a terrible wide spread inhalation hazard. Arsine is really terrible, it will cause
long-term reproductive damage, and cancer concerns, do not tinker with it. It will decompose to its elemental
constituents at around 400°C
providing there is not oxygen present to support its combustion. Electrolysis of solutions that contain
arsenic cations under acid conditions can produce arsine, this is not meant to
be a preparation however, it is a warning.
Boranes: Boron-hydrogen
compound chemistry is extensive, although difficult to facilitate at home. Most of the boranes can be broken down in
some way to the building block molecule borane (BH3), which has
never been isolated on its own, diborane (B2H6) for
example is the dimmer of borane.
Diborane is like all the volatile boranes is toxic, inhalation of
boranes results in headache, dizziness, unconsciousness, fluid in the lungs,
and finally death. Mixes of borane are
spontaneously flammable in moist air, which can cause manipulations involving
it to result in explosions. Boranes
burn with a green flame producing powdery B2O3 that lays
a fine dust on all surroundings. The
main use of diborane is in the preparation of sodium borohydride, a moderately
strong reducing agent that can be safely recrysatalized from  water.
water.
The preparation for boranes is similar to the preparation of silane or of hydrogen sulfide. Acid is slowly dripped over solid magnesium boride (MgB2) resulting in the formation of this spontaneously flammable gas (Note, many boranes are produced, diborane produced is almost instantly hydrolyzed to boric oxide, picture at left shows a milliliter of HCl being dripped onto some magnesium boride). Concentrated phosphoric acid has been shown to give better results in the preparation of boranes. One borane in specific, B4H10, being produced in the largest amounts, this can be converted to diborane by careful distillation at liquid air temperature, under which conditions it dissociates and gives a distillate of diborane.
Being that some boranes are pyrophoric it is easy to assume they might be easily oxidized, that is in this case very true. Atmospheric oxygen and even water will oxidize many of the boranes (though some of the higher anaolges are resistant to oxidation). Therefore all handling vessels must be free of moisture and especially oxygen (however it would be impossible to exclude moisture from the initial flask as it is necessary for the reaction and a part of most acids). Note that diborane was once considered for use as a rocket fuel but the produced boric oxide was too abrasive on the rocket cones, to say this another way, its oxidation is very exothermic and such an exothermic reaction anywhere near you would be disastrous. The industrial process for producing diborane involves the following reaction.
BF3(g) + 6NaH(s)
Þ B2H6(g) + 6NaF(s)
Additionally the reaction between sodium borohydride and elemental iodine in THF or other ethers produces diborane. Although simpler both of these reactions are complicated by the use of two somewhat difficult to obtain chemicals, whereas the magnesium boride used in the first reaction can be theoretically obtained quite readily by a thermite reaction (Note, unless fine reactants are used this reaction is hard to initiate/maintain due to the excess magnesium):
4Mg(s) + B2O3(s)
Þ
3MgO(s) + MgB2(s)
Boric oxide and magnesium shavings being more available then the boron triflouride and sodium hydride used in the industrial process.
Being that boranes are very
toxic gases, any emissions from systems that may contain boranes should be
appropriately dealt with as to reduce their danger. Although these substances can be pyrophoric, one cannot assume that
boranes leaking from a reaction vessel will oxidize before it has a chance to
do any damage to their person.
Therefore one should do either or both of the following; 1) Lower
boranes will react with water to produce boric acid and hydrogen gas readily,
bubbling diborane though a a tall column of NaOH/H2O2,
using an efficient bubbling mechanism (e.g., a glass frit) should greatly
diminish the concentration of diborane.
2) Running the exit gasses of a system into the air intake of a burner. Much care should be taken with boranes, as
many of them are toxic, and potentially explosive, and spontaneously flammable,
this is yet another gas that poses such a danger as to make the author of this
work suggest against working with it.
Butane CH3(CH2)2CH3: Butane is relatively uncreative to may chemical conditions. It makes a workable inert gas for some situations but the actual production of butane on a lab scale from other chemicals is needlessly complicated as it can be purchased for refilling lighters and some camping supplies easily. Butane containing exit gasses should be run into the intake of a torch to prevent butane vapors from accumulating around your work area and causing an explosion hazard. Although butane does not prevent as specific inhalation hazard it can act as an asphyxiant gas, especially if used in an enclosed area.
Carbon Dioxide CO2 WS: Carbon dioxide has no detectable smell. You breath out carbon dioxide and plants
take it in and as such it does not possess some terrible toxicity. But in concentrated form it is hazardous for
your health. Therefore when working
with dry ice or a carbon dioxide cylinder or another source that could provide
a large amount of carbon dioxide quickly into a small space one should take
care and have adequate ventilation.
Aside from commercially available dry ice (solid carbon dioxide) and
cylinders that are available for the soft drink industry, it is also marketed
as a water solution seltzer. Production
of carbon dioxide in the home lab is very simple. The addition of a markedly acid solution to a carbonate or
bicarbonate, either solvated in water or simply as a powder will generate
copious volumes of carbon dioxide, the production of which is controlled by the
rate of acid addition:
CO32-(aq)
+ 2H+(aq) Þ CO2(g) +
H2O(l)
Carbon dioxide produced in this way will contain significant quantities
of water vapor that must be removed using a desiccant and additionally if your
acid has a volatile component (e.g., HCl, HNO3) a portion of the
acid may carry over as well and need its own separate scrubbing. The use of carbon dioxide as an inert
atmosphere is covered in section 8.4 an example of a preparation of carbon
dioxide for lab purposes would entail a setup consisting of a sepretory funnel
on the gas generation flask for the addition of acid at will. The exit gasses for this vessel would be
scrubbed for volatile acid components such as HCl and HNO3 by passing though a
carbonate solution which will in turn make more CO2 and passing those gasses
though strong H2SO4 to remove any water that may be present. A minimum of one trap is required between
the volatile acid scrubbing chamber and the sulfuric acid chamber as suck back
of H2SO4 into saturated carbonate would not be favorable. Additional traps
could be placed though out but the controlled addition of acid to the solution
should ensure that gas continuously flows away from the reaction flask. Carbon dioxide is also the product of
complete combustion but the carbonate method works better for a reasonable
supply. Carbon dioxide is a gas slightly
heavier then air, it possesses no oxidative properties except with strong
reducing agents under heat and it does not act as a reducing agent, upon
dissolution in water it makes a slightly acid solution of carbonic acid.
Carbon Monoxide CO: A colorless, odorless, poisonous gas. It’s toxic action is produced by its strong bonding with the oxygen carrying constituent of human blood, hemoglobin. Carbon monoxide inhalation is treated with oxygen, however since it has no odor, and the usual warning signs of poisoning are lethargy and headache, which can easily be overlooked cases of carbon monoxide poisoning usually go untreated resulting in chronic poisoning or fatal poisoning. The most common reason for carbon monoxide poisoning is from a faulty furnace in the home.
Carbon monoxide is the product of incomplete combustion of carbon containing molecules. It’s use in chemistry is actually quite extensive, however for the beginning chemist it is really not a gas of interest. It does however act as a decent reducing agent, and in environments where carbon is the reducing agent under extreme conditions carbon monoxide can be the main product, therefore taking precautions to remove the gas is an important measure. Other then generating insanely toxic carbonyls of the transition metals though it does not have the wide variety of uses that would render it highly important. It’s preparation is simple, the addition of concentrated sulfuric acid to formic acid results in its dehydration and subsequent generation of carbon monoxide gas.
HCOOH(aq) + H2SO4(l)
Þ
H2SO4*xH2O(aq) + CO(g)
Gas generated in this way is suitable directly for a number of applications provided the addition of the reagents is controlled. The exit gasses of apparatuses that use carbon monoxide or produce carbon monoxide should always be lead into the entry of a flame to burn the gas away entirely. Carbonyl compounds can also be pacified in this manner. However allow me to reiterate the main fact here, carbon monoxide is highly poisonous and gives no warning to its presence, apparatuses using it should be checked and rechecked to ensure they are air tight and exit gasses are properly incinerated, the author of this work recommends against intentionally using carbon monoxide in any preparations.
Chlorine Cl2: (See Section 4.9)
Chlorine Dioxide ClO2 ws: Chlorine
dioxide is surprisingly soluble in water, making a green solution. Its actual reaction with water is slow and
solutions of ClO2 in water are stable for some time. Chlorine dioxide is very toxic and destroys
lung tissue, eye tissue, and skin wholesale upon contact when
concentrated. It is used industrially
for disinfecting and bleaching of paper and as long as the gas is heavily
diluted with an inert gas, usually CO2, it is somewhat safe to handle, it is
usually produced at the point of consumption for this reason though. When concentrated as a gas chlorine dioxide
can explode for no apparent reason or in the presence of traces of organic
material. The usual process to make
ClO2 on the spot involves the reaction between sodium chlorite and sulfuric
acid:
5H2SO4(l)
+ 5NaOCl2(aq) Þ
4ClO2(g) + 2H2O(l) + NaCl(aq) + 2Na2SO4(aq)
Sodium chlorite being the salt of the unstable acid HOCl2. This process is no where near free from danger, the previous method of manufacture of ClO2 was to heat a mixture of sodium chlorate and oxalic acid, this produces CO2 gas simultaneously which dilutes the ClO2 produced, but even with this advantage there have been numerous reports of this preparation going awry and exploding without ascertainable reason. The hydrolysis of ClO2 in water yields a number of products depending on the temperature and time of reaction but the products include chlorate, chlorite, hypochlorite, chloride, and perchlorate. Scrubbing of exit gasses containing ClO2 should be done with strong NaOH, reducing agents should be avoided due to danger of explosion, and apparatuses venting this gasses should be wrapped in screen and shielded from light to lessen the possibility of exploding, exit gasses containing it should never be burned as it may flash back into the system and cause a massive explosion. ClO2 is a very dangerous gas to work with and preparations involving its use and isolation should be avoided.
Ethylene CH2CH2: A colorless gas with a slightly sweet odor. Once used for anesthesia it is exceedingly flammable and explosive under the right conditions. It also has a unique property of acting to ripen fruit despite it being such a simple molecule. Although it can act as an asphyxiant is does not possess any excessive toxic effects on the human body. Unlike acetylene hazardous polymerization is less likely however this gas is quite reactive toward oxidizing agents. Here are a few examples:
CH2CH2(g) + Br2(aq) Þ CH2BrCH2Br(l)
CH2CH2(g) --(KMnO4/NaOH(aq))--> CH2OHCH2OH(aq)
That double bond that ethylene has is quite prone to addition and it is likewise reactive as illustrated above. It has some use in synthesis for such reasons but no use as an inert atmosphere. The production of ethylene is one step further then the production of diethyl ether. In the case of ether two molecules of ethanol are condensed and a water molecule is lost, ethylene goes one step further, another water molecule pulled out of diethyl ether so to speak according to the following reaction:
2CH3CH2OH(l) --(High Heat/Excess H2SO4(l))--> 2CH2CH2(g)
High temperatures (>140 °C) and a significant excess of sulfuric acid (at least a 4x molar excess) to ethanol produce favorable conditions for ethylene formation. It is recommended that you add some fine sand to the reaction mixture to make a slurry that prevents some of the bumping associated with this reaction. As with working with other highly flammable gasses heating should be accomplished by an oil bath held on an electric/non-sparking heating source. The ethylene thus produced after being put though a weak basic wash is suitable for most any purpose.
To scrub ethylene from the exit gasses from a reaction either significant bubbling though an alkaline permanaganate solution or leading the exit gasses into the intake on a flame will work. Although highly flammable, ethylene is less dangerous then some of the gasses mentioned here and can be used with a measure of safety.
Fluorine F2 WS (reacts): (See Section 4.9)
Hydrogen H2: Hydrogen gas is highly flammable although otherwise not terribly reactive. With heating it will form hydrides with some metals and it is a good reducing agent at high temperatures but the kind of run-away reactions that can be expected from some reactions are simply in the lacking with hydrogen (except that whole highly flammable thing). Hydrogen is a very light gas that clears the reaction area very quickly owing to its ability to take flight. The simplicity of making hydrogen gas in the laboratory is only matched by the simplicity of making carbon dioxide. Section 4.2 on acids covers the hydrogen activity series, anything in the list above hydrogen will displace hydrogen from an acid and will therefore produce hydrogen gas as the metal is solvated. If the only acids you have are weak acids you need a somewhat reactive metal, aluminum or magnesium will displace hydrogen from acetic acid (it works with the other metals too but due to dilution the reaction is slow), but usually somewhat stronger acids are available. A great reaction to produce hydrogen gas is to drip hydrochloric acid onto magnesium scrap, the reaction is fast but it is therefore over fast and allows for a more precise control over the speed of hydrogen generation, other good choices might be common steel wool, aluminum foil, or zinc in any form (Do not use nails, you do not want to make a shrapnel bomb!).
Mg(s) + 2HCl(aq)
Þ
MgCl2(aq) + H2(g)
The gas thus produced would have to be scrubbed for two things, volatile acid components (HCL(g)) and for water that vaporizes, it depends on how pure you want this gas as to how much scrubbing you are going to do on it. Running the gas though a sodium hydroxide mixture will eliminate acidic components and running it though a final sulfuric acid wash will remove gas and make the hydrogen suitable for most any application. Exit gasses containing hydrogen are best disposed of by incineration.
Hydrogen Cyanide HCN WS: This is actually a borderline gas/liquid, approx. Bp is 26C
so slightly above room temperature, it has the odor of bitter almonds, however
only 1/3 of the population can smell it due to a genetic defect. But that is not important, what is important
is that this is one of the top four most toxic gasses listed in this
countdown. Not only is it toxic, but it
is prone to explosive polymerization if not properly stabilized and it is also
flammable/explosive on its own.
Hydrogen cyanide is more often then not accidentally made when a chemist
dabbling in cyanides decides to acidify the solution, big mistake:
NaCN(aq)
+ H2O(aq) Û NaOH(aq) + HCN(aq)
I
say big mistake because that equilibrium lies too far to the right for my own
comfort to begin with owing to the weakness of hydrocyanic acid and when acid
is added separately it forces the equilibrium to the right by taking up the
sodium hydroxide and therefore hydrogen cyanide is formed, although very
soluble in water the acid continuously comes acidified solutions, and if
significantly acidified a significant eruption of HCN can occur. Hydrogen cyanide has incredible knockdown
power, one whiff can instantly put a person on the floor unconscious with no
hope of recovery and death following within two or three minutes. Scrubbing hydrogen cyanide from exit gasses
is a simple affair as it is so easily oxidized bubbling though a sodium hypochlorite
solution or other mild to strong oxidizing agent will easily oxidize the
cyanide anion to cyanate OCN- a less toxic version. Hydrogen cyanide really is a chemical that I
recommend others not even work with, as such the information here is more of a
warning then anything amyl nitrate (inhalation) and sodium thiosulfate
(ingestion) are somewhat of folklore medicines to help combat cyanide
poisoning.
Hydrogen Fluoride HF WS: Another borderline
gas/liquid, approximate Bp is 19C, slightly below room temperature. As with all soluble fluorides hydrogen
fluoride would be considered toxic to start out with, however it goes one step
beyond, if you get a splash of hydrogen fluoride on your arm the accepted
scenario involves your flesh dissolving to the bone, then your bone dissolving,
followed by several minutes of pain as you get terrible fluoride poisoning,
finally ending with your heart stopping.
But that’s liquid hydrogen fluoride, nasty stuff anhydrous.
In
dilute solutions (<3%) it is fairly safe to work with, concentrations of
this magnitude are available in over the counter products. They are still toxic but not to the same
degree. As a gas anhydrous hydrogen
fluoride is almost just as bad, eating away at lung tissue and poisoning your
body. At least it’s not flammable. Hydrogen fluoride of all concentrations will
attack glass show in the following equation:
SiO2(s)
+ 6HF(aq) Þ
SiF6H2(aq) + 2H2O(l)
The
ability of hydrofluoric acid to attack glass increases steadily with concentration
to about 99.9%, supposedly totally anhydrous hydrogen fluoride will not attack
glass, however notice that water is created in the attack on glass, therefore
even a stray water molecule could catalyze the reaction. It will eat glass to a significant extent
eliminating the ability to use glass vessels when handling hydrogen fluoride,
either as a solution or as a gas.
CaF2(s)
+ H2SO4(l) Þ CaSO4(s) + 2HF(g)
The
formation of hydrogen fluoride by heating calcium fluoride with concentrated sulfuric
acid is favored by the volatility of the hydrogen fluoride thus formed and
continued heating to keep driving it off.
Additionally hydrogen fluoride could be had by removing the water from
over the counter solutions, either by distillation (be sure to check for
azeotropes) or by dehydration via a strong desiccant.
As
an aqueous solution hydrogen fluoride is a weak acid, this is to be expected if
you look at the trend setup by the other hydrogen halides, but it is actually
much weaker then could be predicted.
This is because the bond between hydrogen and fluorine is incredibly
strong and therefore ionization happens to a considerably lessened extent. It will dissolve many metals though and as
it is doing so the heat of the reaction is probably vaporizing HF out of the
solution and into your air.
Please, hydrogen fluoride is not only a terrible contact poison but
cumulative poison, please consider this information a warning as to its
dangers.
Hydrogen Halides except Fluoride HCl, HBr, HI WS: These are all noxious
smelling gasses at room temperature and pressure. Their solution in water form the recognized acids hydrochloric
acid, hydrobromic acid, and hydroiodic acid.
Inhalation of these gasses can cause damage to the lungs and mucus membranes. Skin contact with concentrated vapors will
result in discoloration and possibly necrosis.
Hydrogen bromide presents a problem different from the other two in that
bromine is not normally utilized by the body, hydrogen bromide inhalation,
resulting in the increase of bromide in the body results in increase lethargy
and in extreme instances, death.
Although not exceedingly toxic these gasses all cause damage on the
physical level, which if enough could cause death.
All
of these gasses are soluble in water to significant extents, hydrogen chloride
the least soluble and hydrogen iodide the most. Their acid solutions provide the most effective ways to generate
the gasses. Dehydration of the
solutions, especially if they are initially concentrated will result in the
formation of the free halogen halide.
Above is an apparatus ideal for the formation of a hydrogen halide gas
from a hydrogen halic acid. In such an
apparatus the acid to be dehydrated is put into 1 which is a funnel closed off
from the rest of the system by the stopcock 2.
This funnel extends downward with a stem that has a very small opening,
almost like capillary tubing 3. This
extends down to the bottom of the vessel and deep under the concentrated
dehydrating acid (sulfuric for hydrogen chloride, phosphoric for bromide or
iodide) 4. The gas upon formation
bubbles though the sulfuric acid and out of the gas tube 5. The opening in the tubing is small enough
that it draws down more acid until the stopcock is closed. This apparatus will generate large
quantities of water-free hydrogen halide.
The simplified version is just a container full of sulfuric acid into
which your hydrohalic acid is dripped in with a sepretory funnel. The disadvantage of this version is the water
spray and evaporation which calls for an additional wash of the exit gasses.
The
hydrogen halides can also be generated from a chemical reaction. Hydrogen chloride can be generated (although
not controllably) by the reaction between sodium chloride and concentrated
sulfuric acid:
NaCl(s)
+ H2SO4(l) Þ NaHSO4(s) + HCl(g)
Although for hydrogen bromide and hydrogen iodide this reaction is not
as feasible. Some of the hydrogen
bromide formed will be oxidized by the sulfuric acid to free bromine and for
the reaction between sodium iodide and sulfuric acid most if not all of the
hydrogen iodide produced is oxidized to iodine. However distillation of weaker solutions of sulfuric acid with
these salts can result in aqueous azeotropes of the acids. The hydrogen halides of these salts can be
generated by the reaction of a sodium salt with concentrated phosphoric acid,
which lacks the oxidizing power to release the free elements.
HBr
and HCl can also be generated as side products from organic halogenations. Usually ½ of the initial halogen is consumed
in the reaction and the other half is released as the hydrogen halide, iodine
however does not posses the power necessary to perform most organic halogenations
and therefore this is not a good way to make hydrogen iodide.
Exit
gasses containing these acidic gasses can easily be scrubbed by bubbling them
though concentrated sodium hydroxide solution.
Subjecting these gasses to high heat will dissociate them to some extent
and oxidizes some of them part way but results in a multitude of products.
Hydrogen Sulfide H2S WS: Yet another incredibly toxic gas, this one more so then
hydrogen cyanide. It has it’s own
unique smell, which everyone has probably smelled at one time or another, the
smell of rotten eggs. However, although
the smell of hydrogen sulfide becomes prominate at levels below lethal levels
it has one major trick, it deadens the sense of smell quickly so the smell goes
away and you think you’re okay, but actually you might just be about ready to
die. Many beginning chemists have died
from hydrogen sulfide thinking that because they could not smell it they were
okay. Smell is only a way of detecting
hydrogen sulfide if it is generated unexpectedly, if that happens leave the
area. You do not want to mess around
with this chemical.
The
treatment of many sulfides with acid is the cause of hydrogen sulfide
production usually. As with hydrogen
cyanide, hydrogen sulfide dissolves in water forming a weak acid solution, and
adding acid to a salt of hydrogen sulfide drives the equilibrium to the
production of hydrogen sulfide gas.
Sulfides can easily be made by the direct combination of an active metal
with elemental sulfur followed by heat.
They can also be the product of high temperature reductions of sulfates.
CaSO4(s)
+ H2(g) Þ
CaS(s) + H2O(g)
S2-(aq)
+ 2H2O(l) Û HS-(aq) + OH-(aq) +H2O(l)
Û H2S(aq)
+ 2OH-(aq)
In
the first equation calcium sulfate is treated with hydrogen at a high
temperature in the absence of oxygen, the products are gaseous water and
calcium sulfide. The second equation
shows the equilibrium that exists in a neutral solution of hydrogen a sulfide salt
of of hydrogen sulfide. Addition of a
base adds hydroxide, which appears on the far right, this drives the equation
to the left, and as long as plenty of base is present sulfides are relatively
safe. However if acid is added that
will destroy the base and it will protonate the sulfide anion floating around
it the solution, both of these will drive the equation to the right and produce
hydrogen sulfide gas.
The
allure of hydrogen sulfide is there though.
It is useful in the laboratory, as an agent to detect certain metal
compounds, as a reducing agent and also to generate some interesting acids.
Br2(l)
+ H2S(g) Þ 2HBr(aq) + S(s)
If
this reaction were to be carried out under a layer of water and stirring were
applied it would not stop there, the sulfur formed would react with the bromine
formatting S2Br2 which would react with the water
resulting in the oxidation of sulfur converting it to SO2 and making
two more molecules of hydrogen bromide, most of which would dissolve in the
upper layer of water making a concentrated hydrobromic acid solution. Solutions of formic, acetic, and other acids
can be made in this manner. Exit gasses containing H2S can be burned
or scrubbed with two washes of concentrated basic solutions or alkali oxidizing
agents such as KMnO4. But
the danger of carrying out such operations usually render this unfeasible,
hydrogen sulfide kills, the author of this work recommends against not working
with this toxic chemical.
Methane CH4: A common gas with which most people are familiar, methane is a simple asphyxiant gas with the side pitfall of being an explosion hazard. It has no uncommon reactivities and behaves fairly inertly. Natural gas piped to homes is mostly methane with other agents added for smell and a small percentage of other hydrocarbons. Methane in home synthesis is somewhat of an extreme measure do to its lack of reactivity. Chlorinating methane should yield carbon tetrachloride, chloroform, methylene chloride, and methyl chloride. However the careful control of temperature and necessary supply of chlorine gas are usually outside of the normal working scope of the home lab. It is interesting to note the simple procedure by which methane can be generated in the home lab:
CH3COONa(s) +
NaOH(s) Þ
CH4(g) + Na2CO3(s)
By simply heating anhydrous sodium acetate with sodium hydroxide the reaction commences generating methane gas and sodium carbonate. Methane can be formed as a product of the electrolysis of some mixtures containing organic components or by the action of water or acid on aluminum or beryllium carbide. If a reaction is run in which methane provides an ‘inert’ atmosphere or a reaction that involves the production or use of methane is run, the exit gasses should be lead into the intake of a burner and incinerated to prevent and explosion hazard.
Nitogen dioxide NO2 WS (reacts): This compound reacts with water to form nitric acid and
nitric oxide gas. Due to this fact it
is incredibly toxic, think about it, it goes into your lungs and makes nitric
acid which in turn causes your lungs to secrete fluid which causes pulmonary
edema which means you’re going to die.
Nitrogen dioxide has a biting odor caused by its hydrolysis upon contact
with fluids within your nose, also due to its hydrolysis you can taste it. It has a high boiling point and can be
easily condensed at home, as a liquid/solid it is colorless in theory due to
the formation of the dimmer N2O4, however it is actually more often then not
brown-red as is the gas.
4HNO3(aq)
+ Cu(s) Þ
2NO2(g) + Cu(NO3)2(aq) + 2H2O(l)
The
easiest production of NO2 involves the action of concentrated nitric
acid upon copper metal. However if the
reason you seek this toxic gas is for the production of nitric acid then that
really does not seem like a feasible method of production. Alternative methods of production include
the moderate temperature decomposition of heavy metal nitrates (although
complicated by the formation of oxygen), or by the action of a strong
electrical discharge on a mixture of oxygen and nitrogen to produce a mixture
of nitrogen oxides followed by a means of separation such as condensation.
6NO2(g)
+ 3H2O(l) Û 3HNO3(aq) + 3HNO2(aq) Û 4HNO3(aq) + 2NO(g) + H2O(l)
As
you can see nitrogen dioxide disproportionates in water to form nitrous acid
and nitric acid, however the nitrous acid thus formed is unstable and
decomposes resulting in the formation of yet another molecule of nitric acid
and a two molecules of nitric oxide.
Giving the overall equation showing that six molecules of nitrogen
dioxide will react with two molecules of water to form four molecules of nitric
acid. It should be noted that nitric
oxide is easily oxidized to nitrogen dioxide by the action of atmospheric
oxygen with no other stimulus.
Solutions of high concentration nitric acid with excess NO2
dissolved within are known as red fuming nitric acid. Incineration of exit gasses containing nitrogen dioxide should be
avoided in combination with any flammable gasses as nitrogen dioxide can
support combustion and cause a flash back of fire into your reaction regardless
of a lack of oxygen.
Exit
gasses containing nitrogen dioxide should be appropriately scrubbed using a
strong sodium hydroxide solution. The
formed sodium nitrate and sodium nitrite may be recovered for future uses by
evaporation of the scrubbing solution afterwards. Nitrogen dioxide is a very poisonous gas, if it is to be used
safety measures should be planned out in advance and it should be used entirely
in a closed reaction system, the operator of such a system should wear a
respirator of some sort and exit gasses should be double washed in concentrated
sodium hydroxide solutions, this is not a gas to take lightly.
Nitrogen N2: Approximately 78 % of the air you breath is, by volume, nitrogen gas. Nitrogen is a diatomic molecule that has a very strong triple bond connecting the two nitrogen atoms. It is inert to a wide variety of common applications except electrostatic discharges. It is purified from air by liquefaction of air followed by fractional distillation. It is a widely available gas in the chemistry industry and can be found for welding applications. Laboratory preparation of nitrogen is very complicated on average, one method being by the action of a strong oxidant on an aqueous ammonia solution, or by the careful heating of ammonium nitrite, or by the careful decomposition of an azide. Other methods also exist but the best is going to be simply removing other components from air. Running normal air into a metal tube full of copper wool and heated externally very hot with a torch will remove the oxygen present if the tube is significantly long enough. You are left with nearly 98% nitrogen content with 1% or so of argon. Nitrogen forms a good inert atmosphere for many reactions. It is an asphyxiant gas like any other, however there is no need to take extra precautions with exit gasses that may contain nitrogen only worry about other things that may be there.
Nitric Oxide (Nitrogen Monoxide) NO: Some references refer to this as nitrogen monoxide, others refer to nitrous oxide as nitrogen monoxide, check the context in older text to be sure which is which. The nitrogen of this molecule has one unpaired electron, which should cause it to at least dimerise forming N2O2, however it does not have a tendency to do this greatly until a low temperature. As a gas nitrogen monoxide is colorless, and somewhat reactive. It will react almost instantly with air forming brown nitrogen dioxide (see above). Nitric oxide is only very slightly soluble in water forming a very small amount of nitrous acid. Nitrogen monoxide can act as either a reducing agent or an oxidizing agent, resulting in the formation of the nitrate anion or nitrogen dioxide respectively.
Nitric oxide is usually formed by the action of dilute nitric acid on copper metal. However it can also be formed by reacting sodium nitrate, ferrous sulfate, and sulfuric acid, which is a convenient laboratory preparation. In all cases the gas must be washed to remove traces of other nitrogen oxides that may be present:
2NaNO2(s) + 2FeSO4(aq)
+ 3H2SO4(aq) Þ 2NO(g) + Fe2(SO4)3(aq)
+ 2NaHSO4(aq) + 2H2O(l)
Solutions of nitrous acid formed by the acidification of a nitrite salt in general will decompose somewhat rapidly to a solution of nitrate and nitrogen monoxide gas:
3NO2-(aq) +
3H+(aq) Û 3HNO2(aq) Þ HNO3(aq) + 2NO(g)
+ H2O(l)
The last step being somewhat irreversible due to the low solubility of NO gas and that it is leaving the solution, therefore driving the reaction to that step. That is one of the reasons why HNO2(aq) is made in situ as needed and not an item to be laying around on a stock shelf. The addition of acid to a nitrite is the way to go, the addition of dilute sulfuric acid to a solution of barium nitrite followed by filtration leading to a somewhat pure solution.
Nitrogen monoxide will support combustion and will attack rubber and possibly lead to an explosion if mixed with potentially oxidizeable gasses. Being that nitrogen monoxide converts to highly poisonous nitrogen dioxide on exposure to atmospheric oxygen your main concern upon venting from a reaction mixture will be the probable inhalation of nitrogen dioxide. However nitrogen monoxide can be scrubbed by passage though a concentrated sodium hydroxide solution, and passing though an open flame may help destroy this molecule, although passage though two or three vats of hydroxide is recommended. As long as you are careful in its use and clean up nitrogen monoxide only poses a hazard to those who disrespect it, if you are working in an area and sudden circumstances cause the evolution of nitrogen monoxide/dioxide gas occur just leave the area, no sense in risking your life.
Nitrous Oxide N2O: Nitrous oxide will readily support the combustion of any of a number of possibly combustible molecules, nearly anything that will burn in air will burn readily in nitrous oxide:
H2(g) + N2O(g)
Þ
H2O(g) + N2(g)
The formation of exceedingly stable dinitrogen being one of the motivating factors in its oxidizing ability. Nitrous oxide has the famous synonym “laughing gas” and is somewhat widely used in the dental profession. It is also used as a foaming agent/propellant for whipped cream due to its high solubility in lipids. Nitrous oxide on its own poses no real danger except the usual asphyxiation hazard, however as stated before it is a potent oxidizing agent therefore its presence in the laboratory should still be monitored and general venting of the gas should be avoided (also in view of its sedative properties).
The classic method of preparation of nitrous oxide is the controlled thermal decomposition of ammonium nitrate:
NH4NO3(s) Þ N2O(g)
+ H2O(g)
During the decomposition extra care must be taken to prevent water from condensing around the top of a flask generating the nitrous oxide and dripping back in, this can increase the production of other nitrogen oxides or may crack a flask. Large amounts of ammonium nitrate should be avoided (>50g) or else the ability of the mass to transfer thermal energy may become inhibited which could result in localized super heating and possibly a run away deflagration. Other safer methods, such as the reaction between hydroxyl amine and sodium nitrate in solution or reacting ammonium nitrate with a small amount of sodium chloride and dilute nitric acid in solution, either one of these reactions requires external heating in a water bath to achieve decent N2O production.
Nitrous oxide should never be produced for human consumption. As an exit gas it should be subjected to thermal decomposition or possibly passed though a bath containing a strong reducing agent.
Nobel Gasses; Helium He/Neon Ne/Argon Ar/Krypton Kr: Helium is readily available mixed with air though party supplies, neon is available though companies catering to neon light production, argon is available to welders, and krypton is a specialty gas. All these gasses are nearly totally unreactive and are good for blanketing a reaction environment. The latter two of them are heavier then air and subsequently do a better blanketing job. As exit gasses these posses the regular asphyxiation hazard but nothing more unless they are carrying with them other potentially hazardous substances which require additional treatment.
Oxygen O2: Making up roughly 20% of the air we breath oxygen is all around us. It’s reactions are numerous but its most noted reaction is a combustion reaction, usually the definitive case between oxygen an a hydrocarbon such as butane:
2CH3(CH2)2CH3(g)
+ 13O2(g) Þ 8CO2(g) + 10H2O(g)
The combustion of such hydrocarbons at least requiring a spark to initiate but once started continuing almost instantly in the case of gasses mixed with oxygen and somewhat more manageably at the liquid gas interface or liquid/gas/solid interface with other forms. If oxygen had been deficient in the above reaction mainly carbon monoxide would have formed in preference to carbon dioxide but only because oxygen had been deficient, carbon monoxide itself will burn if it is mixed with oxygen and the proper spark of activation energy is applied.
Oxygen is a very good and cheap oxidizing agent. Bubbling air though hydrochloric acid with nickel metal immersed in it for example will lead to the dissolution of the nickel somewhat rapidly. At elevated temperatures oxygen attacks many metals, magnesium will readily burn in oxygen as will many finely divided/powdered metals (nickel, iron, zirconium, zinc, etc.). Zirconium powder burning in pure oxygen can achieve temperatures in excess of 4500 °C! Oxygen can also bring out an even higher oxidation state in some elements then even elemental fluorine can. For example, combination of oxygen and osmium metal at room temperature leads to the slow formation of osmium tetroxide which gives osmium a +8 charge, by contrast the reaction of osmium at high temperature with fluorine can only yield the somewhat unstable compound OsF7 (osmium heptafluoride) which decomposes to the more stable osmium hexafluoride.
Dioxygen serves many important oxidizing roles in the lab. But it can also be a nuisance, some materials are so easily oxidized that even a moments exposure to oxygen can contaminate them. A container containing nickel carbonyl most clearly shows this, opening and closing the opening of it for a moment almost immediately causes solid nickel particles to form on the surface of the previously clear solution. Oxygen also leads to explosion hazards and flammability concerns. Liquid oxygen increases these risks, for example, a charcoal briquette soaked in liquid oxygen and then ignited supposedly explodes with the force of a stick of dynamite. However explosives of this type are unpredictable and not commonly used in industry.
Although air contains a relatively decent oxygen percentage it may become necessary to produce oxygen on a lab scale. Purification from air is out of the question for most any amateur chemist due to oxygen being the most reactive component of it, however industrially oxygen is prepared from air by condensing it to a liquid and fractionally distilling off the oxygen (-183 °C). On a home scale oxygen can be prepared by numerous methods, the simplest is the catalytic decomposition of hydrogen peroxide. Adding many different things to hydrogen peroxide will decompose it to oxygen and water. With dilute solutions (<30%) and with monitoring of the temperature (Keeping it less then 70 °C using an ice bath as necessary) this preparation is free of danger. The gas must first be bubbled though concentrated sulfuric acid or another dehydrating agent to remove any water that may be present but afterwards it is of sufficient purity to use. A great catalyst for this is produced by mixing manganese dioxide with sodium silicate until the solution becomes thick then slowly add dilute acid with stirring. The resultant chunks of silica with manganese dioxide imbedded in them are put onto a strainer and washed to remove fine particulates and the granular precipitate is excellent for decomposing peroxide and is fully recoverable.
The classical procedure of making oxygen is the controlled decomposition of a chlorate or perchlorate at medium (150 – 150 °C) temperatures:
2KClO3(s) Þ KClO4(s)
+ O2(g) + KCl(s) Þ 2KCl(s) + 3O2(g)
The decomposition can be stopped at the middle stage producing perchlorate and chloride and oxygen gas or additional or prolonged heating will result in total chloride conversion and maximum production of oxygen gas. The addition of a small amount of manganese dioxide also helps to make the reaction run smoother. However working with nearly molten chlorates/perchlorates is not a nice experience and there is always a chance that the reaction could run out of control. Therefore this method is mentioned for curiosities sake. And also because it is the methodology behind many pyrotechnic mixtures.
Oxygen gas can be safely vented only if there are no additional potentially flammable gasses being released in the vicinity, however if a large quantity of oxygen gas is vented continuously or all at once there is always additional danger associated with it. Exit gasses containing oxygen can be lead into the intake of a burner and will only add to the combustion of the hydrocarbons therein.
Ozone O3: A step up from oxygen in terms of oxidizing ability, ozone will attack may organic compounds with vigor if it comes into contact with them. In very small amounts ozone actually has a pleasant smell, it can be smelt around power lines or when running electrical equipment that can spark such as electric drills. However in higher concentrations the pleasant smell goes away and you are left with a nauseating biting smell. Ozone is capable of innumerable oxidizations, many of which are at least difficult with oxygen and near impossible under the same reaction conditions. For example, ozone will oxidize sulfur dioxide to sulfur trioxide at STP, where as for substantial conversion with oxygen at a minimum an efficient catalyst and somewhat elevated temperatures should be used.
The formation of ozone by chemical means is difficult, especially under controlled circumstances in reliable yields. A number of reactions will produce ozone in small amounts, two such reactions being the oxidation of which phosphorus with oxygen, or the decomposition of manganese heptoxide. However the amounts generated from partial oxidation of phosphorus are small and manganese heptoxide is an unstable explosive liquid.
Electrolysis is another way of making ozone. The electrolysis of perchlorates under alkaline conditions can yield ozone. However the most time tested and reliable way to make ozone is to run oxygen gas though an electric discharge. If nitrogen is present nitrogen oxides may be formed. But a good electric discharge system with a slow steady supply of oxygen flowing though it can yield mixtures of oxygen and ozone containing up to 20% ozone. Ozone is almost always made on site where it is needed and therefore it would be almost impossible to purchase it.
Ozone has its place in chemistry, ozides of the alkali metals can be made with it and extensive oxidations can be carried out with it. It will only be in your exit gas in all likelihood if you put it in your reaction to begin with. If that is the case ozone can be destroyed by bubbling the exit gasses though aqueous solutions of alcohols or reducing agents. High temperatures can destroy ozone but leading it into the intake of a burner will destroy it though combustion. Ozone is highly toxic and as such should be treated with care.
Phosgene COCl2 (Carbonyl Chloride) WS (reacts): Yet another gas who
once found use as a war gas, phosgene is produced industrially from carbon
monoxide and chlorine gas by the simple reaction:
CO(g)
+ Cl2(g) Þ
COCl2(g)
Carbonyl chloride finds use industrially in the
production of polycarbonates. It has a
somewhat high boiling point of 8.7 °C and therefore could be liquefied very easily if that was
your intention. However it is very
toxic to human beings, upon entering the human body it hydrolyzes to
hydrochloric acid and carbon dioxide, the hydrochloric acid attacking the lungs
and eyes causing blindness and pulmonary edema. Its danger should not be
underestimated and any undertaking that could produce phosgene should be
treated with the utmost care and caution.
Phosgene can be produced from the oxidation of chloroform or carbon
tetrachloride. Containers of chloroform
should have ethyl or methyl alcohol added, their presence destroys phosgene by
converting it to hydrochloric acid and ethyl or methyl carbonate
(non-toxic). These chemicals should be
present to the extent of about 1% the total volume, additionally chloroform
should be stored in dark bottles dry and without opening often to limit the
supply of air in the containers.
Exit gasses that contain phosgene should be run
through at least two washes of strongly basified water. Turning it into chloride and carbon
dioxide. Additionally phosgene could be
burned but its combustion would release hydrogen chloride. Addition of an oxidizing agent to the basic
wash is not known to have any additional positive effect.
Phosphine PH3: Unlike
ammonia phosphine only acts as a base in the presence of fairly strong
concentrated acids. Phosphine is only
very slightly water-soluble and a solution of phosphine in water is not
appreciably basic. Phosphine has a
particular smell similar to molded garlic or rancid fish depending on the
individual. The odor is not a very good
indicator as it is toxic below the odor threshold. Phosphine is actually incredibly toxic, and causes terrible
delayed damage to the kidneys and liver.
Similar to arsine poisoning, a person subject to phosphine inhalation
may feel nauseous or ill for a few hours but the symptoms may clear up, only to
have the person struck down in bed again a few days later, with death possibly
following shortly thereafter.
Similar to the production of hydrogen sulfide by
the action of acid on an appropriate sulfide, the action of acid on many
phosphides (of which only zinc phosphine is commonly found) will usually yield
phosphine. An older demonstration for
the properties of phosphine was to take a retort and put some white phosphorus,
water, and sodium hydroxide into it and seal it. Place it on a hot surface and put the beak of the retort so it
dipped just under water. Phosphine
would be produced and bubble up, and as it broke the surface it would
spontaneously ignite and create perfect smoke rings that rose up one after
another. But, just like hydrogen
sulfide, although phosphine is a useful chemical as a reducing agent and for
other reasons, due to its toxicity the author must advise against using
phosphine for any purpose.
The scrubbing of exit gasses containing phosphine can
be accomplished by nearly any oxidizing agent, hypochlorites, permanganate,
ferric salts, even nickel salts will be reduced by phosphine. The most though method though to dispose of
phosphine is to run it into an open flame, this will create a fine dispersion
of phosphorus pentoxide and therefore it is advisable to remain a distance away
from the flame, because although not poisonous it may cause damage to the
throat and lungs.
Propane CH3CH2CH3: Propane follows along the same lines as
the other hydrocarbons in this list (Butane and Methane) in that it is
relatively unreactive to many conditions except oxidation once a certain
activation energy is achieved. It is
widely available at a decent purity and can be used as an inert gas in some
situations. It is an asphyxiant gas and
is flammable so should not be allowed to accumulate. Therefore care should be taken to ignite the exit gasses as they
pass though otherwise they may accumulate.
Silane SiH4: Yet another spontaneously flammable gas at STP when exposed to oxygen:
SiH4(g) + 2O2(g) Þ SiO2(s)
+ 2H2O(g)
Described as having a ‘repulsive odor’ silane has few if any common uses in chemistry. Industrially it is used to produce amorphous silica. Of course that means that when it ignites on its own it will make a cloud of very fine silica which if inhaled will do damage to the lungs, inhalation of fine silica can cause cancer actually. Silane is highly reactive, there are methods in which a chemist could utilize it, for example, reacting it with chlorine would produce silicon tetrachloride, an interesting chemical with a wide variety of uses. But who would want to use such a toxic spontaneously flammable chemical? The normal method of producing silane is to react magnesium silide with acid:
Mg2Si(s) + 4HCl(aq)
Þ
2MgCl2(aq) + SiH4(g)
Mg2Si could in theory be made by direct combination of the elements followed by heating. However it is easiest to produce it in situ by a thermite type reaction between an excess of magnesium and silicon dioxide:
3Mg(s) + 2SiO2(s)
Þ
2MgO(s) + Si(s) + Mg2Si(s)
It is not recommended to use a great excess of magnesium in this mixture, the above equation showing slightly more then is used in practice. If too much magnesium is used, the already difficult to ignite mixture may become impossible to initiate. The mixture thus obtained could be used directly to produce silane.
When present in the exit gasses of a reaction silane can easily be destroyed by running though water, however somewhat basic solutions work even better. The exit gasses may also be destroyed by burning,
Solvent Vapors: In general
solvent vapors should not be vented as most every solvent is highly flammable
with the usual exception of water. As
such the usual method of disposal of solvent vapors is by incineration. The problem with incineration occurs with
halogenated hydrocarbons. Their
incineration entails the production of hydrogen halides which are one of the
gasses specifically described in this test to dispose of in its own way,
incineration not being one of them. If
your exit gasses do contain hydrogen halides it may be good to attempt to take
them out by running them into a beaker full of ice, to re-liquefy them and
therefore not have to deal with their gasses.
They can be burned, but as stated the production of another nuisance gas
is usually not a good reason to burn them.
Many of the more halogenated hydrocarbons will not burn easily though
(Chloroform, carbon tetrachloride).
Although against environmental regulations one may be forced to vent them. However the author of this texts highly
recommends finding another method of dealing with the exit gasses of these.
Sulfur Dioxide SO2 WS: Sulfur dioxide is a useful easily condensed gas with a
boiling point of –10 °C. There are a number
of preparatory methods for this gas including the most obvious of burning
sulfur:
S + O2(g)
Þ
SO2(g)
However this method is difficult to control for a constant source of the gas, as such the addition of oxidizing materials may be necessary to provide a reliable burn rate, in the olden days ‘sulfur candles’ were designed to accomplish the reliable burning of sulfur to sulfur dioxide. In some instances it has been proposed to use some brands of road flares for the preparation of sulfur dioxide but the gasses thus produced usually include some water and carbon dioxide as well. Additionally sulfur dioxide can be prepared from non-combustive methods, notably the reaction between hot concentrated sulfuric acid with copper metal which gives sulfur dioxide and water as the exit gasses. Another notable method involves the reaction between a bisulfite salt (-HSO3) and an acid such as sulfuric or hydrochloric. What this reaction actually does is to drive an equilibrium in the solution toward sulfurous acid which cannot exist in solution in concentrated form, due to this when the concentration increases it leaves the solution as sulfur dioxide. This method is one of the easiest for small amounts of sulfur dioxide to be prepared. Sulfur dioxide is also found in some high-end batteries condensed to a liquid.
Sulfur dioxide is a good reducing agent both in aqueous solution and in other conditions. In aqueous solution sulfurous acid is first formed:
SO2(g) + H2O(l)
Þ
H2SO3(aq)
This acid is easily oxidized to sulfuric acid and in the process reduces a number of compounds, for a simple example, copper salts are reduced to elemental copper. Additionally sulfur dioxide is used in the preparation of sulfuric acid by the contact process, it is passed over heated vanadium pentoxide on a material with a high surface area and sulfur trioxide passes out. This process is difficult to duplicate on a home scale though efforts have been made, additionally some transition metals serve as catalysts in the reaction of sulfur dioxide with water to form sulfuric acid, notably manganese salts though they can only achieve concentrations of ~40% before acid production comes to a stand still.
In addition to being used as a reducing agent and
in the production of sulfuric acid, sulfur dioxide can also be used as a
reagent for the preparation of other reagents, for example, the reaction of
sulfur dioxide with carbonyl chloride at 200 °C (which, may I note is a very
dangerous reaction) can be used for the preparation of thionyl chloride, a very
selective and powerful chlorinating reagent in organic synthesis. There are a number of applications for
sulfur dioxide in the lab, however it is very irritating to the eyes and
throat. Exit gasses containing sulfur
dioxide should be lead through basified water to remove traces of the gas.
Xenon Xe: The heaviest stable noble gas. Xenon has a place of its own due to its incredible reactivity. It will react with a number of very powerful oxidizing agents directly. Although of no consequence to many chemists at home it will react with elemental fluorine, oxides of fluorine, complexes of platinum with oxygen and fluorine, and a few others. But for most everything else it is a great inert gas. For additional properties see the other inert gasses. Xenon is available from specialty gas suppliers.